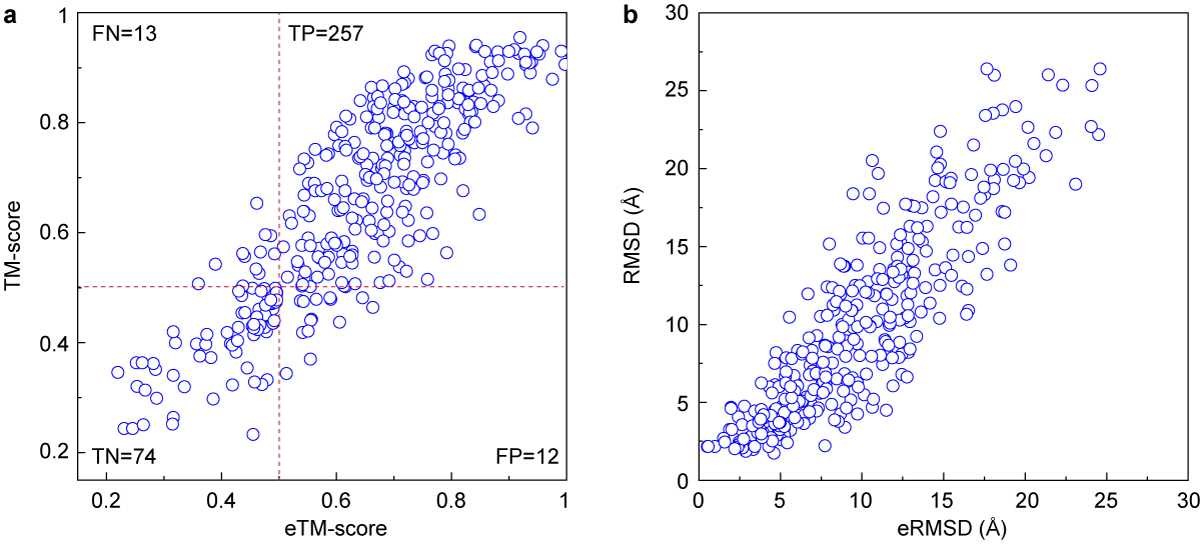
(a)The relationship between the actual TM-score and the eTM-Score of the first model generated by DEMO.
(b)The relationship between the actual RMSD and the eRMSD of the first model generated by DEMO.
TM-score is a scale for measuring the structural similarity between two structures (see Zhang and Skolnick, Scoring function for automated assessment of protein structure template quality, Proteins, 2004 57: 702-710). The purpose of proposing TM-score is to solve the problem of RMSD which is sensitive to the local error. Because RMSD is an average distance of all residue pairs in two structures, a local error (e.g. a misorientation of the tail) will araise a big RMSD value although the global topology is correct. In TM-score, however, the small distance is weighted stronger than the big distance which makes the score insensitive to the local modeling error. A TM-score >0.5 indicates a model of correct topology and a TM-score <0.17 means a random similarity. These cutoff does not depends on the protein length.
You are requested to cite following articles when you use the DEMO server:
1) Xiaogen Zhou, Chunxiang Peng, Wei Zheng, Yang Li, Guijun Zhang, and Yang Zhang. DEMO2: Multidomain protein structures assembly by coupling structural analogous templates with deep-learning inter-domain restraints, to be submitted.
2) Xiaogen Zhou, Jun Hu, Chengxin Zhang, Guijun Zhang, and Yang Zhang. Assembling multidomain protein structures through analogous global structural alignments. Proceedings of the National Academy of Sciences, 116: 15930-15938 (2019).