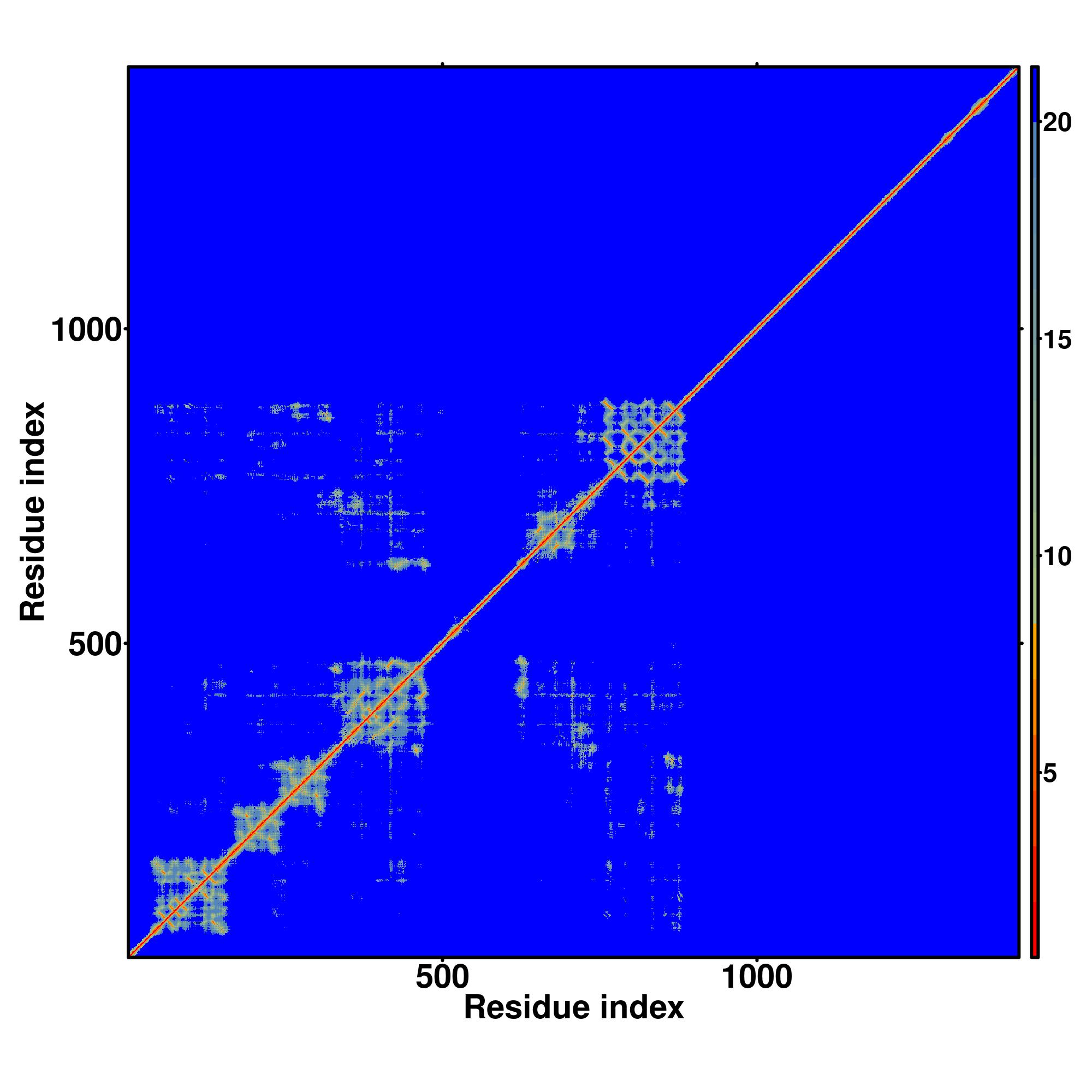
| Rank | PDB
hit | ID1 | ID2 | Cov | Norm.
Zscore | Downloadalignment | | 20 40 60 80 100 120 140 160 180 200
| | | | | | | | | | |
|---|
| SS
Seq | CCCCCCCCCHHHHCHCCCCHHHHHHHHCCHHHHHHHHHCHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHCCCCCCCSCHHHHHHHHHHHCCCCCHHHHHHHHHHHCCCCCCCCSCHHHHHHHHHHHHHHHHHHHHHHHHHCCCCCCCHHHHHHHHHHHHCCCCCCHHHHHHHHHHHCCCHCHCCCCCCCHHHHHHHHCCHHHCCCCCCCCCCCCCCCCC
MSGPWPSPDSRTKGTVAWLAEVLLWVGGSVVLSSEWQLGPLVERCMGAMQEGRRQRTRDQWLKQTFDEADKNGDGSLSIGEVLQLLHKLNVNLPRQRVKQMFREADTDDHQGTLGFEEFCAFYKMMSTRRDLYLLMLTYSNHKDHLDAASLQRFLQVEQKMAGVTLESCQDIIEQFEPCPENKSKGLLGIDGFTNYQERQQRLQGLGRQGPPEEERGTP |
|---|
| 1 | 4tncA | 0.19 | 0.13 | 4.27 | 1.43 | FFAS-3D | | -----------------------------------------MDQQAEARAFL--SEEMIAEFKAAFDMFDADGGGDISTKELGTVMRMLGQNPTKEELDAIIEEVDEDGSGTIDFEEFLVMMVRQMKEEELADCFRIFDKNADGFIDIEELGEILRATG--EHVTEEDIEDLMKDSDKN----NDGRIDFDEFLKMMEGVQ------------------ |
| 2 | 3u0kA2 | 0.17 | 0.13 | 4.19 | 1.21 | SPARKS-K | | -------MVDSSRRKWNKTGHAVRAIGRLSSALT---------------------EEQIAEFKEAFSLFDKDGDGTITTKELGTVMRSLGQNPTEAELQDMINEVDAD-GDGTIDFPEFLIMMAR--KEEEIREAFRVFDDGNGYISAAELRHVMTNLG--EKLTDEEVDEMIREAD----IDGDGQVNYEEFVQMMT--------------------- |
| 3 | 4qoxA | 0.16 | 0.13 | 4.43 | 1.05 | CNFpred | | DLPQFKKISDKAKDLIKKMLM-HEWIKMMT-----QSTQKLAQAALLYMGSKLTTIDETKELTKIFKKMDKNGDGQLDRNELIIGYKELL-AAIEYEVDQILNSIDLDQ-NGYIEYSEFLTVSIDRLSTERLEKAFKLFDKGSGKISANELAQLFGLSD----VSSECWKTVLKEVDQN----NDGEIDFKEFRDMLVKLCNY---------------- |
| 4 | 4i2yA2 | 0.17 | 0.13 | 4.21 | 1.06 | MUSTER | | -----------SRRKWNKAGHAVRAIGRLSSPVVAT--------------RDQLTEEQIAEFKEAFSLFDKDGDGTITTKELGTVMRSLGQNPTEAELQDMINEVDA-DGDGTFDFPEFLTMMARKMNEEEIREAFRVFDDGNGYIGAAELRHVMTDLG--EKLTDEEVDEMIRVADID----GDGQVNYEEFVQMM---------------------- |
| 5 | 4mvfA | 0.16 | 0.14 | 4.68 | 1.13 | HHsearch | | YENDWGSISSDAKNLITKLLEALPWITQMTKSHVE--LSSTLLKNFKKENELHLCDVEINNLRNIFIALDVDNSGTLSSQEILDGLKKIGYQKIPPDIHQVLRDIDSN-ASGQIHYTDFLAATITYLKKEVCLIPFKFFDDGNGKISVEELKRIFGPL------IDKAIDSLLQEVDL----NGDGEIDFHEFMLMMSKLEH----------------- |
| 6 | 4i2yA2 | 0.17 | 0.12 | 4.06 | 1.41 | FFAS-3D | | --------NKAGHAVRA-----IGRLSSPVVATRDQ-----------------LTEEQIAEFKEAFSLFDKDGDGTITTKELGTVMRSLGQNPTEAELQDMINEVDADGDGTFDFPEFLTMMADTDSEEEIREAFRVFDKDGNGYIGAAELRHVMTDLG--EKLTDEEVDEMIRVADID----GDGQVNYEEFVQMM---------------------- |
| 7 | 4i2yA2 | 0.19 | 0.14 | 4.58 | 1.19 | SPARKS-K | | --------SRRKWNKAGHAVRAIGRLSS-PVVATRDQL----------------TEEQIAEFKEAFSLFDKDGDGTITTKELGTVMRSLGQNPTEAELQDMINEVDADG-DGTFDFPEFLTMMNDTDSEEEIREAFRVFDDGNGYIGAAELRHVMTDLG--EKLTDEEVDEMIRVAD----IDGDGQVNYEEFVQMM---------------------- |
| 8 | 3q5iA | 0.14 | 0.11 | 3.87 | 1.04 | CNFpred | | ---------KSDQKTLCGALSNMRKFE---------GSQKLAQAAILFIGSKLTTLEERKELTDIFKKLDKNGDGQLDKKELIEGYNVLR-KNVEEEVDNILKEVDFDK-NGYIEYSEFISVCMILFSEERLRRAFNLFDTKSGKITKEELANLFGLT----SISEKTWNDVLGEADQ----NKDNMIDFDEFVSMMHKIC------------------ |
| 9 | 3u0kA2 | 0.18 | 0.13 | 4.30 | 1.05 | MUSTER | | ------------------------------MVDSSRRKWNKTGHAVRAIGRLSSTEEQIAEFKEAFSLFDKDGDGTITTKELGTVMRSLGQNPTEAELQDMINEVDA-DGDGTIDFPEFLIMMARK--EEEIREAFRVFDDGNGYISAAELRHVMTNLG--EKLTDEEVDEMIREADID----GDGQVNYEEFVQMMT--------------------- |
| 10 | 3q5iA | 0.15 | 0.13 | 4.45 | 1.12 | HHsearch | | DFNDWKNISDEAKELIKLMLEALRWIKKYANNINKSKTLCGALSNMRKFGSKLTTLEERKELTDIFKKLDKNGDGQLDKKELIEGYNVLRLKNVEEEVDNILKEVDFD-KNGYIEYSEFISVCMILFSEERLRRAFNLFDDKSGKITKEELANLFGLTS----ISEKTWNDVLGEADQ----NKDNMIDFDEFVSMMHKIC------------------ |
| (a) | ID1 is the number of template residues identical to query divided by number of aligned residues. |
| (b) | ID2 is the number of template residues identical to query divided by query sequence length. |
| (c) | Cov is equal the number of aligned template residues divided by query sequence length. |
| (d) | Norm. Zscore is the normalized Z-score of the threading alignments. A Normalized Z-score >1 means a good alignment and is highlighted in bold. |
| (e) | Download alignment lists the threading program used to identify the template, and provide the 3D structure of aligned regions of threading templates (threading[1-10].pdb.gz). |
| (f) | Template residues identical to query sequence are highlighted in color. |
|