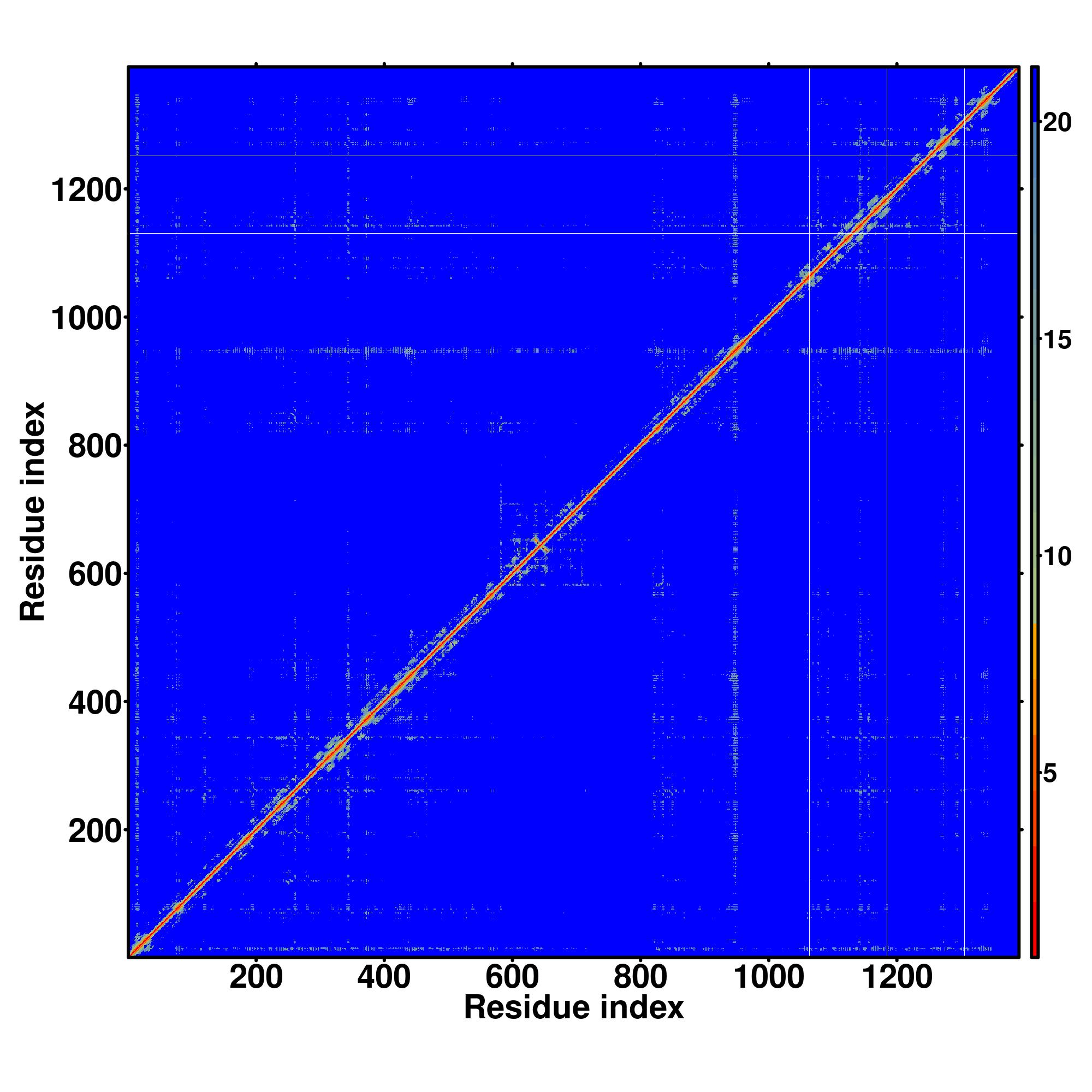
| Rank | PDB
hit | ID1 | ID2 | Cov | Norm.
Zscore | Downloadalignment | | 20 40 60 80 100 120 140 160 180
| | | | | | | | | |
|---|
| SS
Seq | CCCCCCCHHHHHHHHHHHHHHCCCCCCCCCCCCCCCCSSSSSCCCCCCCCCSSSSSSSCCSSSSSSSSSSSSSCCCCCCSSSSSSCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCSSSSSSCCCCCSSSSSSCCCCCSSSSSSCCCCCCCSSCCCCCSSSCCCCCCCCCCCCCSSCCCCCSSSSSCCCCSSSSC
MKAPAVLAPGILVLLFTLVQRSNGECKEALAKSEMNVNMKYQLPNFTAETPIQNVILHEHHIFLGATNYIYVLNEEDLQKVAEYKTGPVLEHPDCFPCQDCSSKANLSGGVWKDNINMALVVDTYYDDQLISCGSVNRGTCQRHVFPHNHTADIQSEVHCIFSPQIEEPSQCPDCVVSALGAKVLSSVKDRFINFF |
|---|
| 1 | 2eceA | 0.07 | 0.06 | 2.36 | 1.00 | DEthreader | | ------------------HFGWNACSSAGCGPDAILRKRKRIHSLTLGENRMALELRPLLMGFINMVSSIWLWFYEKWNAEKVIEIPAEP------L--E-GNLPELKPFKAVPPLVTDIDISL-DDKFLYLSLWG-IGEVRQYDISNFKPVLT-GKVKLGGHRADKLTGAPYVTNSLYTWDSHQVRLSGGDASS- |
| 2 | 4fwwA | 0.33 | 0.26 | 7.66 | 1.99 | SPARKS-K | | --------------------------------------VKYVVPSFSAGGLVQAMVTYEGAVFVAIRNRLHVLGP-DLKSVQSLATGPAGD-PGCQTCAACGPGPHGPPG---DTDTKVLVLDP-ALPALVSCGSSLQGRCFLHDLEPQGTAVHLAAPACLFSAHHNRPDDCPDCVASPLGTRVTVVEQGQASYFY |
| 3 | 1olzA | 0.13 | 0.09 | 3.21 | 0.63 | MapAlign | | -------------------------------------------VQFHEIYNYSALLLSKDTLYIGAREAVFAVNALNIEKQHEVYWKVSEDK--KAKCAEK----GKSKQTECLNYIRVLQPL--SATSLYVCGTAFQPACDHLNLT--SFKFLGKNEDGCPFDPAHSYTSYSGTSYNFLGSEPISRNSSHSPLRT |
| 4 | 4fwwA | 0.33 | 0.26 | 7.66 | 0.54 | CEthreader | | --------------------------------------VKYVVPSFSAGGLVQAMVTYESAVFVAIRNRLHVLGP-DLKSVQSLATGPAGD-PGCQTCAACGPGPH---GPPGDTDTKVLVLDPAL-PALVSCGSSLQGRCFLHDLEPQGTAVHLAAPACLFSAHHNRPDDCPDCVASPLGTRVTVVEQGQASYFY |
| 5 | 4fwwA | 0.33 | 0.26 | 7.66 | 1.41 | MUSTER | | --------------------------------------VKYVVPSFSAGGLVQAMVTYEGAVFVAIRNRLHVLGP-DLKSVQSLATGPAGD-PGCQTCAACGPG---PHGPPGDTDTKVLVLDP-ALPALVSCGSSLQGRCFLHDLEPQGTAVHLAAPACLFSAHHNRPDDCPDCVASPLGTRVTVVEQGQASYFY |
| 6 | 4fwwA | 0.32 | 0.25 | 7.53 | 2.71 | HHsearch | | --------------------------------------VKYVVPSFSAGGLVQAMVTYESAVFVAIRNRLHVLGP-DLKSVQSLATGPAGD-PGCQTCAACGPGPHG---PPGDTDTKVLVLDP-ALPALVSCGSSLQGRCFLHDLEPQGTAVHLAAPACLFSAHHNRPDDCPDCVASPLGTRVTVVEQGQASYFY |
| 7 | 4fwwA1 | 0.32 | 0.22 | 6.77 | 1.50 | FFAS-3D | | ----------------------------------------------------QAMVTYESAVFVAIRNRLHVLGP-DLKSVQSLATGPAGDPG----CQTCAACGPGPHGPPGDTDTKVLVLDP-ALPALVSCGSSLQGRCFLHDLEPQGTAVHLAAPACLFSAHHNRPDDCPDCVASPLGTRVTVVEQGQASYFY |
| 8 | 1q47B1 | 0.10 | 0.08 | 2.99 | 0.70 | EigenThreader | | --------------------------------KNNVPRLKLSVITFNGSSSYHTFLLDESRLYVGAKDHIFFNLV-NIKDFQKIVWPVS--YTRRDECKWA----GKDILKECANFIKVLEAY--NQTHLYACGTAFHPICTYIEVGIFKLQDSH-FENGRGKSPYDPKLLTDVVMFIGVGTVLKVVSLEEVLLEE |
| 9 | 1shyB | 1.00 | 0.80 | 22.43 | 3.31 | CNFpred | | ---------------------------------------KYQLPNFTAETPIQNVILHEHHIFLGATNYIYVLNEEDLQKVAEYKTGPVLEHPDCFPCQDCSSKANLSGGVWKDNINMALVVDTYYDDQLISCGSVNRGTCQRHVFPHNHTADIQSEVHCIFSPQIEEPSQCPDCVVSALGAKVLSSVKDRFINFF |
| 10 | 5f30A | 0.09 | 0.07 | 2.76 | 1.00 | DEthreader | | -DTCPIMH--HM--AAF--P--------S-PDPYKEFYDNLQRDAAEGLGLGVHVTITADGYAVGDGDICAEFDRETDMVRYAWAFDWDPNV---AWLDGG--ELIEDG-PLHSVANDALVFDPR-GKWAVASMRL-PGVCVVFDRE--NQVPVAV-LAGPKISANRQNNIMVW------NTFHMVFTPDERISTT |
| (a) | ID1 is the number of template residues identical to query divided by number of aligned residues. |
| (b) | ID2 is the number of template residues identical to query divided by query sequence length. |
| (c) | Cov is equal the number of aligned template residues divided by query sequence length. |
| (d) | Norm. Zscore is the normalized Z-score of the threading alignments. A Normalized Z-score >1 means a good alignment and is highlighted in bold. |
| (e) | Download alignment lists the threading program used to identify the template, and provide the 3D structure of aligned regions of threading templates (threading[1-10].pdb.gz). |
| (f) | Template residues identical to query sequence are highlighted in color. |
|