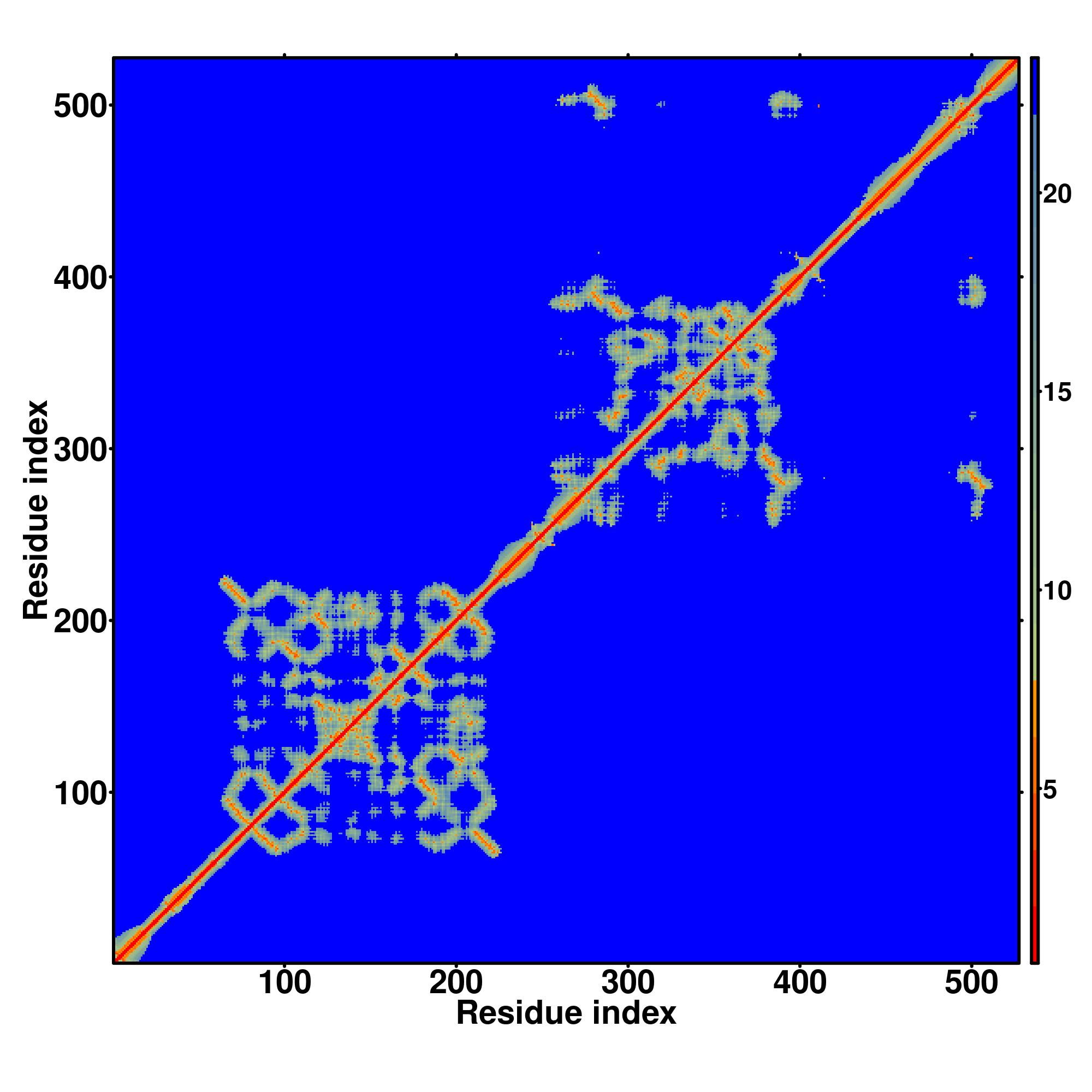
| Rank | PDB
hit | ID1 | ID2 | Cov | Norm.
Zscore | Downloadalignment | | 20 40 60 80 100 120 140 160 180 200
| | | | | | | | | | |
|---|
| SS
Seq | CCCHHHHHHHHHHHHHHHCCCCCCHHHCCCCHHHHHHHHHHHHHHHHHHHHHHCCSSCCCCCCSSSSSCCCCCCCCCCCCCCCCCCSSSSCCCCCCCCCCCSSSCCSSCCCCCCCCCCCSSSSSSCCCCCCCCCCCCCCCCCCCCCCCSSCCCHHHCCCCCCCCCSSCCCCCCCCCCCSSSSSSCCCCCCCCCCCCCCSSSSCCCCCCCCSSSSSSSCC
MNSPCDRLQQFIQVLLEESWSFPSFANTLHWPENLLSYIDELVWQGSLQNFHQHEVRFDKPPLRLPLTGFSSLTENWSSRQAVSSRLVATAASPPAGCQAPIAFLGLKFSSLGPARKNPALCFLYDQSNSKCNTSWVKENVGCPWHWCNIHEALIRTEKGSDPMFYVNTSTGGRDGFNGFNLQISDPWDPRWASGVDGGLYEHKTFMYPVAKIRIARTL |
|---|
| 1 | 1lcsA1 | 0.13 | 0.08 | 2.68 | 1.07 | CEthreader | | --------------------------------------------------------------PHQVYNVTWTITNL------VTGTKANATSMLGTLTDAFPTMYF------------PFYVCPGHANRKQC---GGPQDGFCAVWGC---ETTGETYWRPTS----SWDYITVKKGVPLILQFTQKGRQTWDGPKSWGLRLYRSGYDPIALFSVSRQV |
| 2 | 1aolA | 0.11 | 0.07 | 2.63 | 1.13 | MapAlign | | ----------------------------------------------------------------QVYNITWEVTNGDR-------ETVWAISGNHPTWWPVLTPDLLRDQVTHKSSEGFYVCPGSHREAKSC---GGPDSFYCASWGC-ETTGRVPSSSWDYITVDNQAVQVCKDNKWCNPLAIQFKQVTSWTTGHYWGLRLYVSGRDPGLTFGIRLRY |
| 3 | 1lcsA1 | 0.12 | 0.07 | 2.43 | 1.05 | HHsearch | | --------------------------------------------------------------PHQVYNVTWTITNLVT--GTKANATSMLG---------TLTFPTMYF--------PFYVCPGHANR-KQCGG---PQDGFCAVWGCETTGETYWRPTS--S-----WDYITVKKGVPLILQFTQKGRTSWDGPKSWGLRLYRSGYDPIALFSVSRQV |
| 4 | 1aolA | 0.09 | 0.09 | 3.30 | 0.66 | CEthreader | | ---------QVYNITWEVTNGDRETVWAISGNHPLWTWWPVLTPDLCMLALSGPPHWGLEYQAPYSSPPGPPCCSGSSGSSAGCSRDCDEPLTSLTPRCNTAWNRLKLDQVTHKSSEGFYVCPGSHRPREA-KSCGGPDSFYCASWGCETTGRVYWKPSSSWDYITVDNNLTTSQAVQVCKDNKWCNPVTSWTTGHYWGLRLYVSGRDPGLTFGIRLRY |
| 5 | 1lcsA | 0.09 | 0.06 | 2.40 | 0.67 | EigenThreader | | --------------------------------------------------------------PHQVYNVTWTITNL---VT---GTKANATSMLTDAFPT-MYFDLCDIIGNTWNPGCPFYVCPGHANRKQCGGPQD--GFCAVWGCETTGETYWRPTSSWDQGIGPCYTGASEGGRCNPLILQFTQKGRQTSWPKSWGLRLYRSGYDPIALFSVSRQV |
| 6 | 1ltlE | 0.16 | 0.14 | 4.71 | 0.39 | FFAS-3D | | MKTVLTKFEEFVFEAIEKYPNVPDLADLLEKPDDVIRAAQQAIRNIRLRKNVDLNIRFSGISNVIPLREFVAVDGIVRKTDEIRPRIVKAVEC--RGCMRHHAVTQSTNMITEPSL-----C---SECGGRSFRLLQDESEFLDTQTLKLQEPLENLSGGEQPR--------------QITVVLEDDLVDTLTPGDIGTLRTVRDERTKRFKNFIYGN- |
| 7 | 1lcsA1 | 0.16 | 0.09 | 3.05 | 0.67 | SPARKS-K | | --------------------------------------------------------------PHQVYNVTWTITNLVTGTKANATSMLGTLDAFP----------TMYFP--------FYVCPG-HANRKQCG---GPQDGFCAVWGCE---TTGETYWRPTSSWDYITVKKGVP----LILQFTQKGRQTWDGPKSWGLRLYRSGYDPIALFSVSRQV |
| 8 | 5m4sA | 0.11 | 0.05 | 1.64 | 0.51 | CNFpred | | NTTLGNSLQESLDELIQSQQITPQLALQ--VLLQFDKAINAALAQRVRNRVNFGSLNTYR---FCDNVWTFVLNDVEVREELIKVDKVKIVACD----------------------------------------------------------------------------------------------------------------------------- |
| 9 | 1jk9B | 0.07 | 0.05 | 2.26 | 0.83 | DEthreader | | --ACLKNVPINSLNFDIQQIM---S--VESSVA-PSTIINTLRN-CGKD--A---------GAGKPNSSAVAILETFQKYTKDTAVRGLARIVQVGENKTL-FD-ITVNGVP--EAGNYHASIHE-K-----G--DVSKGVETGK--VWHKFDE------------PIE--CFNESYSGKTFLSAPLPTWQLI-GRSFVISKSLHPEDYSFLGVIARSA |
| 10 | 1lcsA | 0.09 | 0.06 | 2.27 | 0.87 | MapAlign | | -----------------------------------------------------------------HQVYNVTWTITNLV--TGTKANATSMLGTLTDAFPTMYFDLCDIRRWQQRNTPFYVCPGHANR-KQC---GGPQDGFCAVWGC-ETTGETPTSSWDYITVKKGVTQGISEGGRCNPLILQFGRQTSWDGPKSWGLRLYRYDPIALFSVSRQVMT |
| (a) | ID1 is the number of template residues identical to query divided by number of aligned residues. |
| (b) | ID2 is the number of template residues identical to query divided by query sequence length. |
| (c) | Cov is equal the number of aligned template residues divided by query sequence length. |
| (d) | Norm. Zscore is the normalized Z-score of the threading alignments. A Normalized Z-score >1 means a good alignment and is highlighted in bold. |
| (e) | Download alignment lists the threading program used to identify the template, and provide the 3D structure of aligned regions of threading templates (threading[1-10].pdb.gz). |
| (f) | Template residues identical to query sequence are highlighted in color. |
|