| Rank | PDB
hit | ID1 | ID2 | Cov | Norm.
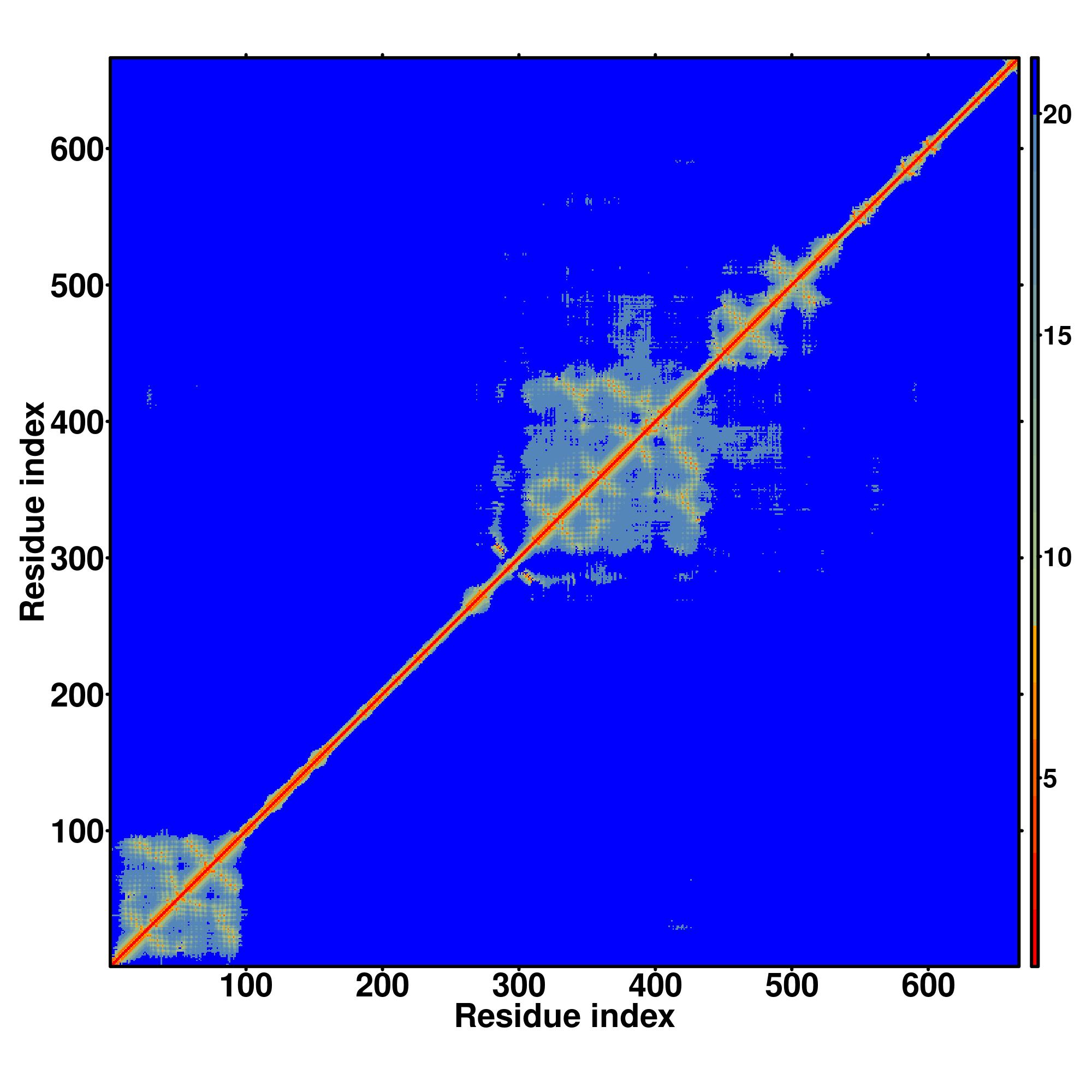
Zscore | Downloadalignment | | 20 40 60 80 100 120 140 160 180 200 220 240 260 280 300 320 340 360 380 400
| | | | | | | | | | | | | | | | | | | | |
|---|
| SS
Seq | CCCCCCCCCCHHHHCCCCCCCCCCCCCCCCCCCCCCCCCCCCCHHHHHHHHCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCHHHCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCHHHHHHHHHHCCCCCHHHHCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCHHHHHHHHHHHHHHCCCCCHHHHHHHHHHHCCCCCHHHHHHHHHHHCCHHHHHHHHHHHHHHHHHHHHHHHHCCCCCCCCCCCCCCCCCCCCCHHHHHHCCHHHHHHHHHHHHHHHHHHCCCCCCCCCHHHHHCCCCCCHHHHHHHHHHHHHHHCCCHHHHHHHHHHHHHHHCCHHHHHHHHHCCCCCCCCCCCHHHHHHHHCCCC
DAPGSCLIDCNEKTRKKSQKETESLHCEYVAEPVMAQSTQNVDYNQLQEVIYPETLKLEGKGPELVGPSESKPRGTSPLPAGQVPVTLQPQKQVKENKTQPPVAYQYWPPAELQYRPPPESQYGYPGMPPAPQGRAPYPQPPTRRLNPTAPPSRQGSELHEIIDKSRKEGDTEAWQFPVTLEPMPPGEGAQEGEPPTVEARYKSFSIKMLKDMKEGVKQYGPNSPYMRTLLDSIAYGHRLIPYDWEILAKSSLSPSQFLQFKTWWIDGVQEQVRRNRAANPPVNIDADQLLGIGQNWSTISQQALMQNEAIEQVRAICLRAWEKIQDPGSACPSFNTVRQGSKEPYPDFVARLQDVAQKSIADEKARKVIVELMAYENANPECQSAIKPLKGKVPAGSDVISEYVKACDGIG |
|---|
| 1 | 4o9xA | 0.06 | 0.04 | 1.79 | 0.67 | DEthreader | | --------ITGMGEAL-TPTGGMAALSLPLPISAYAPAF---------DCNVMTIGPEGSHFSDQSQIYYQYRAETAQRYLH-I-VYYG----SETLP-L----------LVFDYGERSSTTGS------RQILNYDSTLLGSGQGD--GLGYRSSSQFWLDEKAAA-LTTGQTPVYARGAWDG------------REREFRGFGY-VEQTD-------------LT-KNW--YA---TGLPVIDNALSTYWRDDQAFAGFS--PR--FT--TWQ-DNK----DVPLTP----EDDN----SRYWFNRALKGQRSQVSSVVESRNYCSQYPRR-----------PDTLPD-LL--S---YDPIGREIKVRRTVNEDPTDFHRT-YDPVGNV----------------S---I |
| 2 | 6ssjA | 0.97 | 0.52 | 14.63 | 2.09 | SPARKS-K | | ---------------------------------------------------------------------------------------------------------------------------------------------------------------------------------PV--------TLEPV------EARYKSFSIKMLKDMKEGVKQYGPNSPYMRTLLDSIAHGHRLIPYDWEILAKSSLSPSQFLQFKTWWIDGVQEQVRRNRAANPPVNIDADQLLGIGQNWSTISQQALMQNEAIEQVRAICLRAWEKIQDPGSACPSFNTVRQGSKEPYPDFVARLQDVAQKSIADEKARKVIVELMAYENANPECQSAIKPLKGKVPAGSDVISEYVKACDGIG |
| 3 | 6ssjA | 0.97 | 0.52 | 14.56 | 1.13 | MapAlign | | -----------------------------------------------------------------------------------------------------------------------------------------------------------------------------------PVTLE------------PVEARYKSFSIKMLKDMKEGVKQYGPNSPYMRTLLDSIAHGHRLIPYDWEILAKSSLSPSQFLQFKTWWIDGVQEQVRRNRAANPPVNIDADQLLGIGQNWSTISQQALMQNEAIEQVRAICLRAWEKIQDPGSACPSFNTVRQGSKEPYPDFVARLQDVAQKSIADEKARKVIVELMAYENANPECQSAIKPLKGKVPAGSDVISEYVKACDGIG |
| 4 | 6ssjA | 0.97 | 0.52 | 14.56 | 0.92 | CEthreader | | -----------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------PVTLEPVEARYKSFSIKMLKDMKEGVKQYGPNSPYMRTLLDSIAHGHRLIPYDWEILAKSSLSPSQFLQFKTWWIDGVQEQVRRNRAANPPVNIDADQLLGIGQNWSTISQQALMQNEAIEQVRAICLRAWEKIQDPGSACPSFNTVRQGSKEPYPDFVARLQDVAQKSIADEKARKVIVELMAYENANPECQSAIKPLKGKVPAGSDVISEYVKACDGIG |
| 5 | 6ssjA | 0.97 | 0.52 | 14.56 | 1.39 | MUSTER | | ----------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------PVTLE-PVEARYKSFSIKMLKDMKEGVKQYGPNSPYMRTLLDSIAHGHRLIPYDWEILAKSSLSPSQFLQFKTWWIDGVQEQVRRNRAANPPVNIDADQLLGIGQNWSTISQQALMQNEAIEQVRAICLRAWEKIQDPGSACPSFNTVRQGSKEPYPDFVARLQDVAQKSIADEKARKVIVELMAYENANPECQSAIKPLKGKVPAGSDVISEYVKACDGIG |
| 6 | 6ssjA | 0.99 | 0.53 | 14.89 | 3.63 | HHsearch | | ---------------------------------------------------------------------------------------------------------------------------------------------------------------------------------PVTLE----P----------VEARYKSFSIKMLKDMKEGVKQYGPNSPYMRTLLDSIAHGHRLIPYDWEILAKSSLSPSQFLQFKTWWIDGVQEQVRRNRAANPPVNIDADQLLGIGQNWSTISQQALMQNEAIEQVRAICLRAWEKIQDPGSACPSFNTVRQGSKEPYPDFVARLQDVAQKSIADEKARKVIVELMAYENANPECQSAIKPLKGKVPAGSDVISEYVKACDGIG |
| 7 | 6ssjA | 0.99 | 0.53 | 14.82 | 2.12 | FFAS-3D | | ---------------------------------------------------------------------------------------------------------------------------------------------------------------------------------PVTL--------------EPVEARYKSFSIKMLKDMKEGVKQYGPNSPYMRTLLDSIAHGHRLIPYDWEILAKSSLSPSQFLQFKTWWIDGVQEQVRRNRAANPPVNIDADQLLGIGQNWSTISQQALMQNEAIEQVRAICLRAWEKIQDPGSACPSFNTVRQGSKEPYPDFVARLQDVAQKSIADEKARKVIVELMAYENANPECQSAIKPLKGKVPAGSDVISEYVKACDGIG |
| 8 | 6ssjA | 0.99 | 0.53 | 14.89 | 0.98 | EigenThreader | | ---------------------------------------------------------------------------------------------------------------------------------------------------------------------------------PVTLEPV--------------EARYKSFSIKMLKDMKEGVKQYGPNSPYMRTLLDSIAHGHRLIPYDWEILAKSSLSPSQFLQFKTWWIDGVQEQVRRNRAANPPVNIDADQLLGIGQNWSTISQQALMQNEAIEQVRAICLRAWEKIQDPGSACPSFNTVRQGSKEPYPDFVARLQDVAQKSIADEKARKVIVELMAYENANPECQSAIKPLKGKVPAGSDVISEYVKACDGIG |
| 9 | 6ssjA | 1.00 | 0.53 | 14.95 | 1.83 | CNFpred | | ---------------------------------------------------------------------------------------------------------------------------------------------------------------------------------PVTLEP--------------VEARYKSFSIKMLKDMKEGVKQYGPNSPYMRTLLDSIAHGHRLIPYDWEILAKSSLSPSQFLQFKTWWIDGVQEQVRRNRAANPPVNIDADQLLGIGQNWSTISQQALMQNEAIEQVRAICLRAWEKIQDPGSACPSFNTVRQGSKEPYPDFVARLQDVAQKSIADEKARKVIVELMAYENANPECQSAIKPLKGKVPAGSDVISEYVKACDGIG |
| 10 | 4iglA | 0.07 | 0.05 | 1.97 | 0.67 | DEthreader | | --------INGMGESV-GQAGGMVTFSIPLPFSAVAPAL---------CSAMSI-SPSGQDFSPDQTHILGHSRVSGENQRYLARIDY------ASLF-L----------LVFDYGERDEGGTTG-----HQTLDYDVSTGSGQQG--DGLGYRSSAQFWLDEKQLV-EAAGQQPECAHGSWDG------------QEREFRGFGR--LIQR-----------SRTV--SW--FA---TGIPEIDTTLSAEFWRDDQAFSPF--SP--RF--TR-WEDS----DVAFIP----S-E-----HDAFWLNRAMKGQLRHQSTIETRSYQYDPQCR---PDDLPETL-------------HIYDPLGREYQVITRRERVNQDE--ND-------------------TA--AN--- |
| (a) | ID1 is the number of template residues identical to query divided by number of aligned residues. |
| (b) | ID2 is the number of template residues identical to query divided by query sequence length. |
| (c) | Cov is equal the number of aligned template residues divided by query sequence length. |
| (d) | Norm. Zscore is the normalized Z-score of the threading alignments. A Normalized Z-score >1 means a good alignment and is highlighted in bold. |
| (e) | Download alignment lists the threading program used to identify the template, and provide the 3D structure of aligned regions of threading templates (threading[1-10].pdb.gz). |
| (f) | Template residues identical to query sequence are highlighted in color. |
|