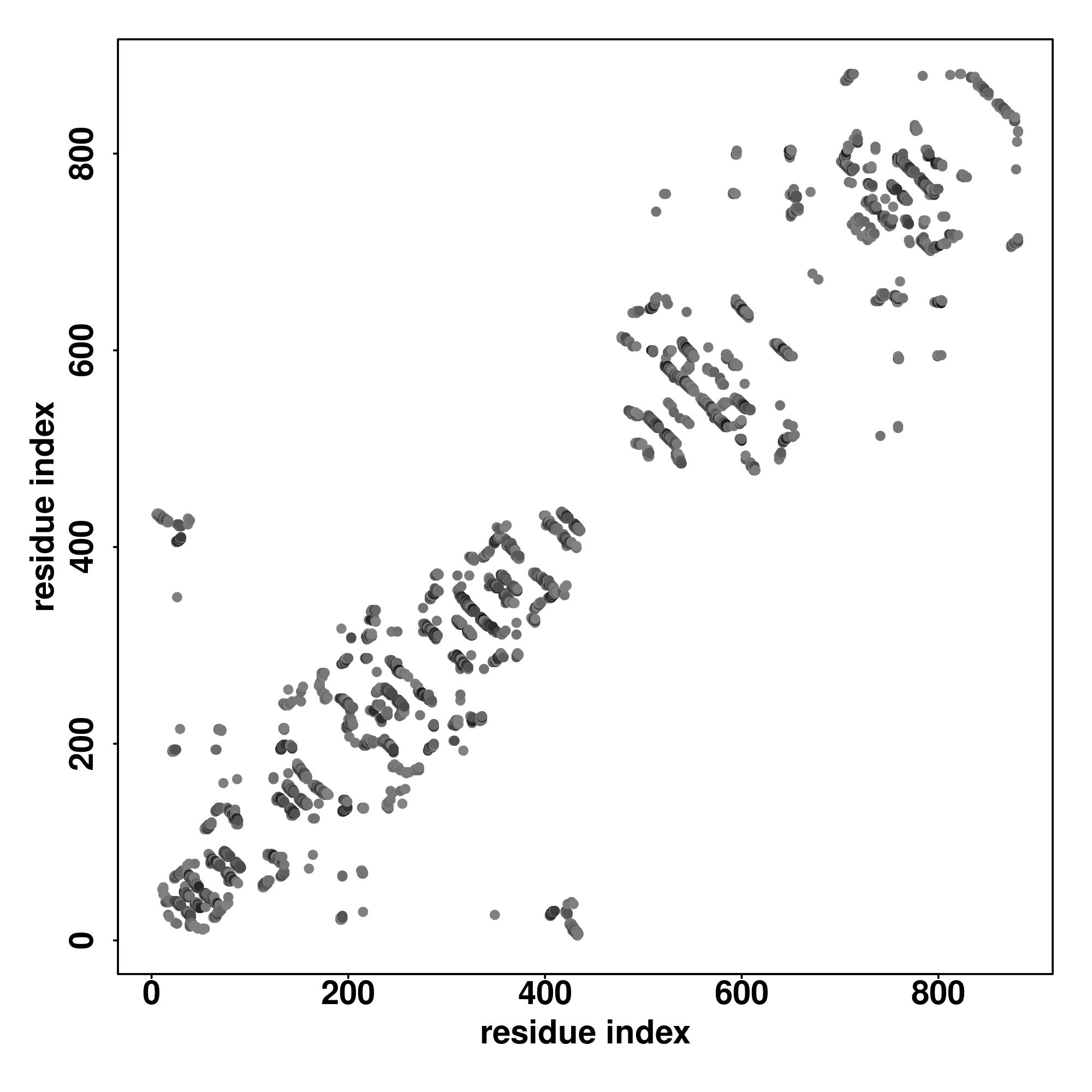
| Rank | PDB
hit | ID1 | ID2 | Cov | Norm.
Zscore | Downloadalignment | | 20 40 60 80 100 120 140 160 180 200 220 240
| | | | | | | | | | | | |
|---|
| SS
Seq | CCCCHHHCCCSSSSSCCCCSSSSSSSSSSCCCCCCCCCCCCHHHHHHHCCCCSSSSSHHHCCCCCCCCCCCCCCCCCCCCCCSSSSCCCCCSSSSSSCCCCCCCCCCCSSSSCCCCCSSSSSSCCCCSSSSSCCCCSSSSSSCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCSSSSSSSCCCCCCCCSSSSSSCCCCSSSSSSCCCCCCCCCCCCCCHHHHHHHHHHHHHHHHHHHHHHHHHHCC
GADATLLQSPVVLCGLPDGQLCCVILKALVTSRSAPGDPNALVKILHHLEEPVIFIGALKTEPQAAEAAENFLPDEDVHCDCLVAFGHHGRMLAIKASWDESGKLVPELREYCLPGPVLCAACGGGGRVYHSTPSDLCVVDLSRGSTPLGPEQPEEGPGGLPPMLCPASLNICSVVSLSASPRTHEGGTKLLALSAKGRLMTCSLDLDSEMPGPARMTTESAGQKIKELLSGIGNISERVSFLKKA |
|---|
| 1 | 3j80g2 | 0.13 | 0.08 | 2.83 | 0.38 | CEthreader | | PKNLVDDGRITFVSAGMDKIVRSWSLNS-----------YRIEADFIGHNNYINVVQPS------------------PDGSLAASAGKDGQIYVWNLK------HKSAFMNFDAKDEVFALAFSSRFWLTAATASGIKIYDL--------------ENEVLIDELKPEFAGYPHAVSLAWS----ADGQTLFAGYTDNVIRVWQVMTAN------------------------------------- |
| 2 | 6rlbC | 0.07 | 0.07 | 2.64 | 0.62 | EigenThreader | | VTCCCLSKAFLLFAGTAHGSVVVWDLRELHYSVTLFWTFRTATFSTVNHRSPLQAVEPISLSPF------STQEEMSGLSFHIASLDESGVLNVWVSISDLGLMPGGRVKLVHSALTLNVKFLSDPNHFIIGTDMGISHGTRQDLRVAPKLPQ-------QHGIRPVKVNVIDFSFLAGCSDGSTDSHAVTGLQWSPAVFLVQDDTSSGSLKRRWAAPEVDECNRLRLLLQ--------------- |
| 3 | 6vbu91 | 0.15 | 0.14 | 4.77 | 0.60 | FFAS-3D | | -VDNTGNGQDKIIVGSFMGYLRIFPILQVEVGKFVSGTEMLHLAVLH---SRKLCVYSVSGTLGNVEHGNQYSFGGVKGRDLICIQSVDGMLMVFEQ---ESYAFGRFLPGSLLPGPL--AYSSRTDSFITVSSCQVESYKYQVLAFAVVDWTLNIGEQAIDICIVSFIQSASSVFVLGERNFKDNGQIQIVTLSDDGHLQCSYLGTDPSKVESRELNYDELDMELKELQKVIKNVNK-------- |
| 4 | 3i2nA | 0.16 | 0.13 | 4.34 | 0.85 | SPARKS-K | | GGLGIGEGAPEIVTGSRDGTVKVWDPVANMEPVQGENKRDCWTVAFGNAEERVVCAGYLFDLRNMALRWETNIKNKDISMNKLVATSLEGKFHVFDMRTQHPTKGFASVSEKAHKSTVWQVRHLQNRELFLTAGGALHLWKYEYPIQRKDSEGIEMGVAGSVSLLQNVTLSTQPISSLDWS---PDKRGLCVCSSFDQTVRVLIVTK--------------------------------------- |
| 5 | 3acpA | 0.11 | 0.07 | 2.50 | 1.60 | CNFpred | | ---------KYVIAGHVSGVITVHNVFSK----------EQTIQLPSKFTCSCNSLTVDG-----------------NNANYIYAGYENGMLAQWDLRS----PECPVGEFLIN-TPINNVYFAAGALFVSSGFDTSIKLDII-----SDPESERPAIE---ETPTFLVSNDDEVSQFCYVSD--ESNGEVLEVGKNNFCALYNLSNP-------------------------------------- |
| 6 | 1l5gA | 0.10 | 0.08 | 2.86 | 1.00 | DEthreader | | SKDFTAVDDFVSGVPRARTLGMVYIYDGKN--------M-SSLYNFTGEQAYFGFSVAATDIN------------GDDYADVFIGAPLVGQVSVSLQR-A---SGDFQTTKLNGFEAFSAIAPLGDNDIAIAAYGIVYIFN-GR--ST----------GLNAVPSQILEGQWPSFYSMKGATDIKNGYPDLIVGAFVDRAILYRARPVPILNQFTPA-----------------GVVRNNEALLAG |
| 7 | 6jp6A2 | 0.10 | 0.07 | 2.56 | 0.45 | MapAlign | | ----SQSGDFLLVTVYSDSTIKIWHY--------RENQNKFDLIMQGRYTCCLFNVVFIALK----------------EELLVVISPTDGHLVVYNISVDISGDLVLPVAQLPVHSGVKSLDYVASATILTGGDNGLGLSNLKL---------DDSNKVTLKTSDFIAAAASSTITSGMLING----GKEVITTSVDQVIRAWEITAGKLS----------------------------------- |
| 8 | 5oqld | 0.12 | 0.11 | 3.87 | 0.54 | MUSTER | | EGGPSLSFHRTFAASSGQGCIRIWDLEHSTAGQAQWPSFVDTITDVCFNQVETSVIGSVATDRSLRTNMPVIKTVLHFACNRIVFNPMEAMNLAVASEARNFDKALNIQKGH--VAAVMDVEFSPTGEELVSGSYD--TIRL------------RRDAGHSRDVYHTKRMQRVFRTMWT-----MDSK-YILTGSDDGNVRLWRAN-ASERSGVKATRQRQALEYNNALLDRYGHL-PEIRRIRRH |
| 9 | 2oajA2 | 0.13 | 0.10 | 3.38 | 0.52 | HHsearch | | KTAINNSNIGFVGIAYAAGSLMLIDRRGPAIIYMEIREIS-G-----AQSACVTCIEFVIMEYGD----------DGYSSILMVCGTDMGEVITYKIL-PASGKFDVQLMDITSKGPIHKIDAFSKGIVLITGFDDIRLITLGKSKST----------HK----GF-KY-PLAATGLISTVNNDRKNLTVIITLEINGHLRVFTIPD-FKEQMSEHIP---------------------------- |
| 10 | 3dwlH | 0.11 | 0.08 | 2.95 | 0.36 | CEthreader | | LSLDWHPNNVLLAAGCADRKAYVLSAYVRDVDKPEASLPFNTVCAEYPSGGWVHAVGFSPSG------------------NALAYAGHDSSVTIAYPSAPEQPPR-ALITVKLSQLPLRSLLWANESAIVAAGYYSPILLQGNEAGTSKTSFTHTGNTGEGREESSLPTVHQNMIATLRPYAGTPGNITAFTSSGTDGRVVLWTL----------------------------------------- |
| (a) | ID1 is the number of template residues identical to query divided by number of aligned residues. |
| (b) | ID2 is the number of template residues identical to query divided by query sequence length. |
| (c) | Cov is equal the number of aligned template residues divided by query sequence length. |
| (d) | Norm. Zscore is the normalized Z-score of the threading alignments. A Normalized Z-score >1 means a good alignment and is highlighted in bold. |
| (e) | Download alignment lists the threading program used to identify the template, and provide the 3D structure of aligned regions of threading templates (threading[1-10].pdb.gz). |
| (f) | Template residues identical to query sequence are highlighted in color. |
|