| Rank | PDB
hit | ID1 | ID2 | Cov | Norm.
Zscore | Downloadalignment | | 20 40 60 80 100 120 140 160 180 200 220 240
| | | | | | | | | | | | |
|---|
| SS
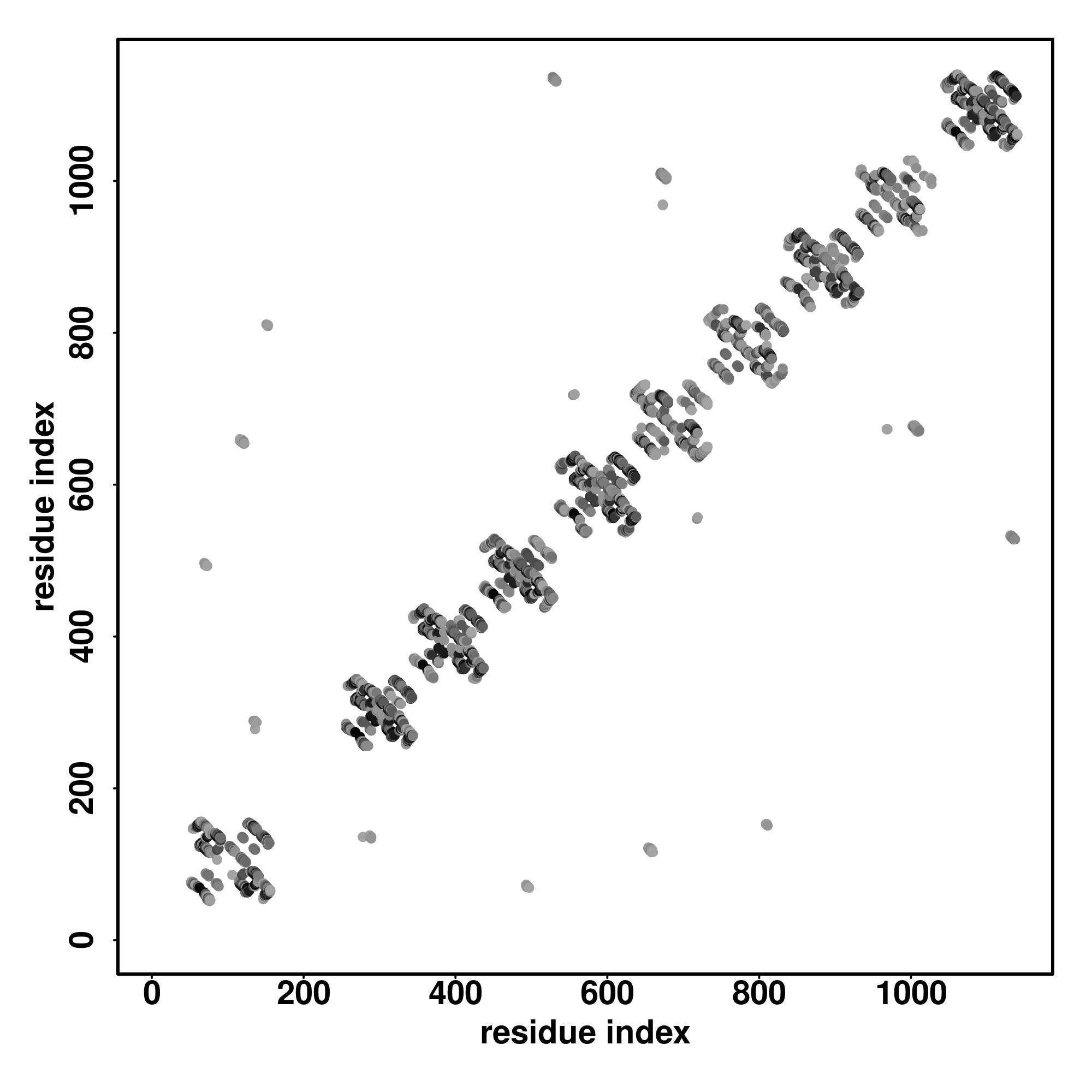
Seq | CCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCSSSSCCCCSSSSCCCSSSSSSSSSCCCCCCCCSSSSSSCCSSSCCCCCCCSSSSCCCCCCCCCCCSSSSSSCCCCHHHCSSSSSSSSCCCCSSSSSSSSSSSCCCCCCCCCCCCCCSSSSCCCCCSSSSCCCSSSSSSSSCCCCCSSSSSSCCSSCCCCCCSSSSSCCCSSSSSSCCCCHHHCSSSSSSSSSSSSSSSSSSSC
MPEAKPAAKKAPKGKDAPKGAPKEAPPKEAPAEAPKEAPPEDQSPTAEEPTGVFLKKPDSVSVETGKDAVVVAKVNGKELPDKPTIKWFKGKWLELGSKSGARFSFKESHNSASNVYTVELHIGKVVLGDRGYYRLEVKAKDTCDSCGFNIDVEAPRQDASVEVKKSAAFTKKLDPAYQVDRGNKIKLMVEISDPDLTLKWFKNGQEIKPSSKYVFENVGKKRILTINKCTLADDAAYEVAVKDEKCFTELFVKE |
|---|
| 1 | 1ya5A | 0.22 | 0.16 | 5.09 | 1.36 | SPARKS-K | | ----------------------------------------------MTTQAPTFTQPLQSVVVLEGSTATFEAHISGFP---VPEVSWFRDGQVISTSTL-------PGVQISFSDGRAKLTIPAVTKANSGRYSLKATNGSGQATSTAELLVKAET--------APPNFVQRLQSM-TVRQGSQVRLQVRVTIPTPVVKFYRDGAEIQSSLDFQISQEGDLYSLLIAEAYPEDSGTYSVNATNSTSTAELLVQG |
| 2 | 1ya5A | 0.22 | 0.16 | 5.09 | 1.25 | MUSTER | | ----------------------------------------------MTTQAPTFTQPLQSVVVLEGSTATFEAHISGFP---VPEVSWFRDGQVISTSTLPG-------VQISFSDGRAKLTIPAVTKANSGRYSLKATNGSGQATSTAELLVKA--------ETAPPNFVQRLQS-MTVRQGSQVRLQVRVTIPTPVVKFYRDGAEIQSSLDFQISQEGDLYSLLIAEAYPEDSGTYSVNATNSTSTAELLVQG |
| 3 | 1ya5A | 0.21 | 0.15 | 4.87 | 2.46 | FFAS-3D | | ----------------------------------------------MTTQAPTFTQPLQSVVVLEGSTATFEAHISGFP---VPEVSWFRDGQVISTSTLPGVQISFSD-------GRAKLTIPAVTKANSGRYSLKATNGSGQATSTAELLVKAETAPPNFVQR---------LQSMTVRQGSQVRLQVRVTIPTPVVKFYRDGAEIQSSLDFQISQEGDLYSLLIAEAYPEDSGTYSVNATNSVGTAELLVQG |
| 4 | 2rikA | 0.27 | 0.19 | 5.91 | 2.88 | CNFpred | | ------------------------------------------------MAPPFFDLKPVSVDLALGESGTFKCHVTGT---APIKITWAKDNREIRP---GGNYKMTLV------ENTATLTVLKVTKGDAGQYTCYASNVAGKDSCSAQLGVQEP-----------PRFIKKLEPSRIVKQDEHTRYECKIGGPEIKVLWYKDETEIQESSKFRMSFVESVAVLEMYNLSVEDSGDYTCEAHSASSSTSLKVKE |
| 5 | 7b17B | 0.10 | 0.07 | 2.49 | 0.83 | DEthreader | | -------------------------------------------------Q--VQLVETGGGLVQPGGSLRLSCAASG-FTFSSYAMGWARQLEWVSYQDSVKRFTISRDNA----KSTVYLQMNSLKPEDTAVYYCATESLGDGSWGQGTQVTVSS------------QV-QLVETGGGFVGSLRLSCAASG---VTLDYAIGWCIGSDGRISRDNAK----NTVYL-QMNSLDAYYCALTVG-DYWGKGTQVTV |
| 6 | 6fwxB | 0.22 | 0.16 | 4.96 | 1.35 | SPARKS-K | | --------------------------------------------------PPNFVQRLQSMTVRQGSQVRLQVRVTGIP---TPVVKFYRDGAEIQSSLD---FQISQEG------DLYSLLIAEAYPEDSGTYSVNATNSVGRATSTAELLVQGETT-------QAPTFTQPLQS-VVVLEGSTATFEAHISFPVPEVSWFRDGQVIDSSPGVQISFSDGRAKLTIPAVTKANSGRYSLKATNGTSTAELLVKA |
| 7 | 3b43A | 0.17 | 0.15 | 5.01 | 0.42 | MapAlign | | ----------ASLVINKVDHSDVGEYTCKAENSVGAVASSAVLVIKERKLPPSFARKLKDVHETLGFPVAFECRINGS---EPLQVSWYKDGEL-LKD--DANLQTSF------IHNVATLQILQTDQSHVGQYNCSASNPLGTASSSAKLTLSEH--------EVPPFFDLKPV-SVDLALGESGTFKCHVTGTPIKITWAKDNREIRPGGNYKMTLVENTATLTVLKVTKGDAGQYTCYASNDSCSAQLGVQE |
| 8 | 4u7mA | 0.16 | 0.13 | 4.16 | 0.46 | CEthreader | | --------------------------------------------------KPQIITQPETTMAMVGKDIRFTCSAASSS-SSPMTFAWKKDNEVLTNAD-----MENFVHVHAVMEYTTILHLRQVTFGHEGRYQCVITNHFGSTYSHARLTVNVLPSFTKTPHLETPSLVVPL-EDRVVSVGETVALQCKATGPPPRITWFKGDRPLSLTERHHLTPD--NQLLVVQNVVAEDAGRYTCEMSNERAHSQLSVLL |
| 9 | 6fwxB | 0.21 | 0.15 | 4.75 | 1.20 | MUSTER | | --------------------------------------------------PPNFVQRLQSMTVRQGSQVRLQVRVTGIP---TPVVKFYRDGAEIQSSLD---------FQISQEGDLYSLLIAEAYPEDSGTYSVNATNSVGRATSTAELLVQGETT-------QAPTFTQPLQS-VVVLEGSTATFEAHISFPVPEVSWFRDGQVISSGPGVQISFSDGRAKLTIPAVTKANSGRYSLKATNGTSTAELLVKA |
| 10 | 3b43A | 0.23 | 0.20 | 6.41 | 0.47 | HHsearch | | GVQEPPRFIIVKQDEHTRYECKIGGSPEQESSDAHNAAGSASSSTSKVKEPPVFRKKPHPVETLKGADVHLECELQGTP---PFQVSWHKDKRELRS---GKKYKIM------SENFLTSIHILNVDSADIGEYQCKASNDVGSDTCVGSITLKAP-----------PRFVKKLSD-ISTVVGEEVQLQATIEAEPISVAWFKDKGEIVREDNIWISYSENIATLQFSRAEPANAGKYTCQIKNEECFATLSVLE |
| (a) | ID1 is the number of template residues identical to query divided by number of aligned residues. |
| (b) | ID2 is the number of template residues identical to query divided by query sequence length. |
| (c) | Cov is equal the number of aligned template residues divided by query sequence length. |
| (d) | Norm. Zscore is the normalized Z-score of the threading alignments. A Normalized Z-score >1 means a good alignment and is highlighted in bold. |
| (e) | Download alignment lists the threading program used to identify the template, and provide the 3D structure of aligned regions of threading templates (threading[1-10].pdb.gz). |
| (f) | Template residues identical to query sequence are highlighted in color. |
|