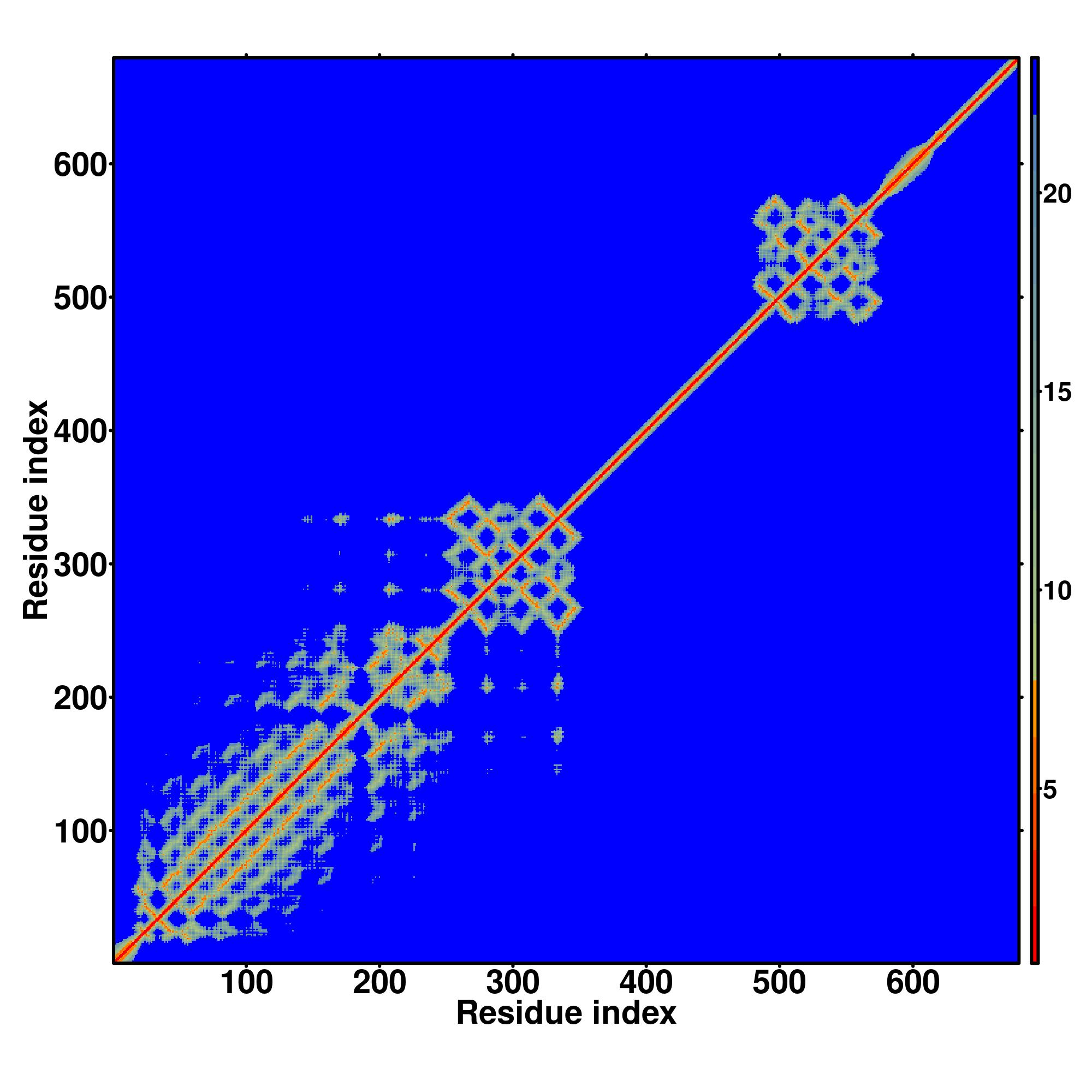
| Rank | PDB
hit | ID1 | ID2 | Cov | Norm.
Zscore | Downloadalignment | | 20 40 60 80 100 120 140 160 180 200 220 240
| | | | | | | | | | | | |
|---|
| SS
Seq | CHHHHHHHHHHHHHCCCCCCCCCCCSSCCCCCCCCCCCCSSSCCCCCCCCCCCCCCCCCCSSSCCCCCCCSSCHHHCCCCCCCCSSSCCCCCCCSCCCCCCCCCCCCCSSSCCCCCCCCCCCCCCCCCCCCCSSSCCCCCCCSSCHHHHCCCCCCCSSSCCCCCCCSCCHHHHCCCCCCCCCCCHHHCCCCCCCSSSSCCCCCCCCCCHHHHHHHHHHHCCCCCCCCCCCSCCCCCCCCCCSCCCCCHHHCCCC
MHLFACLCIVLSFLEGVGCLCPSQCTCDYHGRNDGSGSRLVLCNDMDMNELPTNLPVDTVKLRIEKTVIRRISAEAFYYLVELQYLWVTYNSVASIDPSSFYNLKQLHELRLDGNSLAAFPWASLLDMPLLRTLDLHNNKITSVPNEALRYLKNLAYLDLSSNRLTTLPPDFLESWTHLVSTPSGVLDLSPSRIILGLQDNPWFCDCHISKMIELSKVVDPAIVLLDPLMTCSEPERLTGILFQRAELEHCLKP |
|---|
| 1 | 4rcaB | 0.24 | 0.21 | 6.49 | 1.33 | DEthreader | | ----------------VCKEK-I--CSCNEIEGDLSQFYHLFLHGNSLTRLFEFAFYNAVSLHMENNGLHEIVPGAFLGLQLVKRLHINNNKIKSFRKQTFLGLDDLEYLQADFNLLRDIDPGAFQDLNKLEVLILNDNLISTLPANVFQYV-PITHLDLRGNRLKTLPYEVLEQIPGI--------------AEILLEDNPWDCTCDLLSLKEWLENIPKNALIGR--VVCEAPTRLQGKDLNETTEQDLCP- |
| 2 | 3wo9A | 0.29 | 0.25 | 7.56 | 2.60 | SPARKS-K | | ------------HMACLAVGKDDICTCSNKT---DSSPETVDCSSKKLTAVPTGIPANTEKLQLDFNQLANIPAEAFHGLTRLTYLALDYNQLQSLPVGVFDQLNNLNELRLQDNQLTSLPPGVFDSLTKLTYLTLSQNQLQSIPAGVFDKLTNLNRLELSTNQLQSVPHGAFDSLVNLE--------------TLHLELNPWDCACSIIYLRTFIAKNTDKISGME-SAQCN----GTSTAVKDVNTEKIKNV |
| 3 | 2v9tB | 0.27 | 0.22 | 6.77 | 0.55 | MapAlign | | -------------------HCPAACTCS---------NNIVDCRGKGLTEIPTNLPETITEIRLEQNTIKVIPPGAFSPYKKLRRIDLSNNQISELAPDAFQGLRSLNSLVLYGNKITELPKSLFEGLFSLQLLLLNANKINCLRVDAFQDLHNLNLLSLYDNKLQTIAKGTFSPLRA--------------IQTMHLAQNPFICDCHLKWLADYLHT---NPIETS-GARCTSPRRLANKRIGQIKSKKFRC- |
| 4 | 2v9tB | 0.27 | 0.22 | 6.78 | 0.41 | CEthreader | | ----------------GSLHCPAACTCSN---------NIVDCRGKGLTEIPTNLPETITEIRLEQNTIKVIPPGAFSPYKKLRRIDLSNNQISELAPDAFQGLRSLNSLVLYGNKITELPKSLFEGLFSLQLLLLNANKINCLRVDAFQDLHNLNLLSLYDNKLQTIAKGTFSPLRAI--------------QTMHLAQNPFICDCHLKWLADYLHTNP----IETSGARCTSPRRLANKRIGQIKSKKFRC- |
| 5 | 3wo9A | 0.28 | 0.24 | 7.46 | 2.01 | MUSTER | | ------------HMACLAVGKDDICTCSNKT---DSSPETVDCSSKKLTAVPTGIPANTEKLQLDFNQLANIPAEAFHGLTRLTYLALDYNQLQSLPVGVFDQLNNLNELRLQDNQLTSLPPGVFDSLTKLTYLTLSQNQLQSIPAGVFDKLTNLNRLELSTNQLQSVPHGAFDSLVNLE--------------TLHLELNPWDCACSIIYLRTFIAKNTDKISGM-ESAQCNGT----STAVKDVNTEKIKNT |
| 6 | 5xnpA | 0.30 | 0.27 | 8.12 | 1.00 | HHsearch | | --------------PGDPQICPKRCVCQILS-----PNLATLCAKKGLLFVPPNIDRRTVELRLADNFVTNIKRKDFANMTSLVDLTLSRNTISFITPHAFADLRNLRALHLNSNRLTKITNDMFSGLSNLHHLILNNNQLTLISSTAFDDVFALEELDLSYNNLETIPWDAVEKMVSLHTLSKGTFSHSPSTFALSFGGNPLHCNCELLWLRRLSRE--------DDLETCASPPLLTGRYFWSIPEFLCEPP |
| 7 | 2v9tB | 0.28 | 0.22 | 6.87 | 2.04 | FFAS-3D | | -----------------SLHCPAACTCS---------NNIVDCRGKGLTEIPTNLPETITEIRLEQNTIKVIPPGAFSPYKKLRRIDLSNNQISELAPDAFQGLRSLNSLVLYGNKITELPKSLFEGLFSLQLLLLNANKINCLRVDAFQDLHNLNLLSLYDNKLQTIAKGTFSPLRAIQ--------------TMHLAQNPFICDCHLKWLADYLHTNPIE----TSGARCTSPRRLANKRIGQIKSKKC--- |
| 8 | 4rcaB | 0.19 | 0.17 | 5.46 | 0.77 | EigenThreader | | ------------------DVCKEKICSC----NEIEGDLHVDCEKKGFTSLQRFAPTSQFYHLFLHNSLTRLFPNEFANFYNAVSLHMENNGLHEIVPGAFLGLQLVKRLHINNNKIKSFRKQTFLGLDDLEYLQADFNLLRDIDPGAFQDLNKLEVLILNDNLISTLPANVFQYVPITTLPYEEVLEQIPGIAEILLEDNPWTCD--LLSLKEWLENIPKNALIGRVVCEATRLQ---GKDLNETTEQDLCP- |
| 9 | 2v9sA | 0.27 | 0.22 | 6.76 | 6.06 | CNFpred | | -------------------HCPAACTCSN---------NIVDCRGKGLTEIPTNLPETITEIRLEQNTIKVIPPGAFSPYKKLRRIDLSNNQISELAPDAFQGLRSLNSLVLYGNKITELPKSLFEGLFSLQLLLLNANKINMLRVDAFQDLHNLNLLSLYDNKLQTIAKGTFSPLRAI--------------QTMHLAQNPFICDCHLKWLADYLHTN----PIETSGARCTSPRRLANKRIGQIKSKKFR-- |
| 10 | 2v9tB | 0.28 | 0.23 | 6.99 | 1.33 | DEthreader | | --------------G--SLHCPAACTC-S--------NNIVDCRGKGLTEIPTNLPETITEIRLEQNTIKVIPPGAFSPYKKLRRIDLSNNQISELAPDAFQGLRSLNSLVLYGNKITELPKSLFEGLFSLQLLLLNANKINCLRVDAFQDLHNLNLLSLYDNKLQTIAKGTFSPLRAI--------------QTMHLAQNPFICDCHLKWLADYLHTN-P-IETSG--ARCTSPRRLANKRIGQIKSKKFR-C |
| (a) | ID1 is the number of template residues identical to query divided by number of aligned residues. |
| (b) | ID2 is the number of template residues identical to query divided by query sequence length. |
| (c) | Cov is equal the number of aligned template residues divided by query sequence length. |
| (d) | Norm. Zscore is the normalized Z-score of the threading alignments. A Normalized Z-score >1 means a good alignment and is highlighted in bold. |
| (e) | Download alignment lists the threading program used to identify the template, and provide the 3D structure of aligned regions of threading templates (threading[1-10].pdb.gz). |
| (f) | Template residues identical to query sequence are highlighted in color. |
|