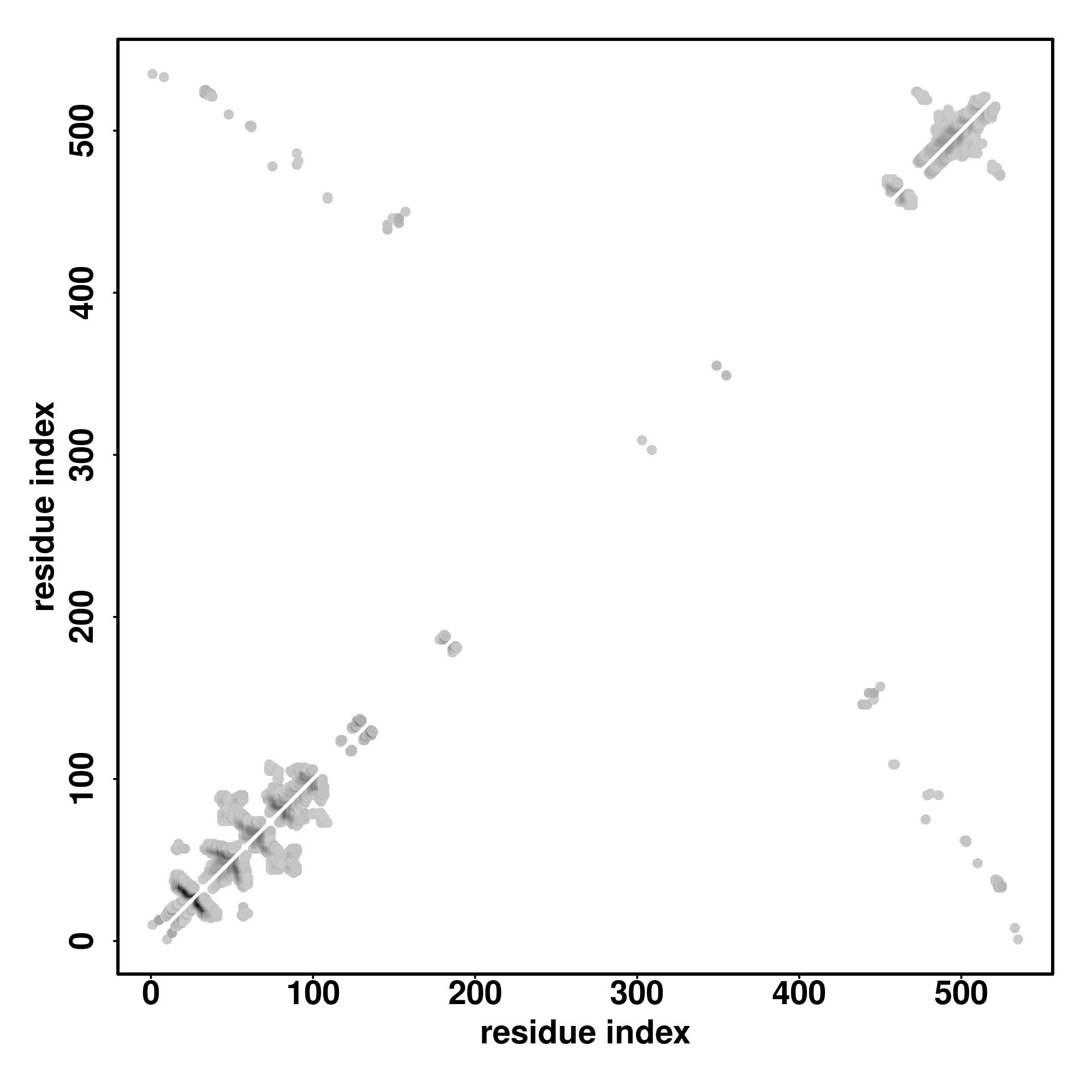
| Rank | PDB
hit | ID1 | ID2 | Cov | Norm.
Zscore | Downloadalignment | | 20 40 60 80 100 120 140 160 180 200 220 240 260 280 300 320 340 360 380 400
| | | | | | | | | | | | | | | | | | | | |
|---|
| SS
Seq | CCCHHHCCHHHHHCCHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHCCCCCCCCCCHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHCCCCSSSSCCCCCCCCHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHCCCCCCCCCSSSSCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCSSSCCCCCCC
TVNQSLLTPLHVEIDPEIQRVRTQEREQIKTLNNKFASFIDKVRFLEQQNKVLETKWALLQEQGQNLGVTRNNLEPLFEAYLGSMRSTLDRLQSERGRLDSELRNVQDLVEDFKNKYEDEINKHTAAENEFVVLKKDVDAAYMGRMDLHGKVGTLTQEIDFLQQLYEMELSQVQTHVSNTNVVLSMDNNRNLDLDSIIAEVKAQYELIAQRSRAEAEAWYQTKYEELQVTAGKHGDNLRDTKNEIAELTRTIQRLQGEADAAKKQCQQLQTAIAEAEQRGELALKDAQKKLGDLDVALHQAKEDLTRLLRDYQELMNVKLALDVEIATYRKLLESEESRMSGECPSAVSISVTGNSTTVCGGGAASFGGGISLGGSGGATKGGFSTNVGYSTVKGGPVSAGTSILRKTTTVKTSSQRY |
|---|
| 1 | 7kogB | 0.17 | 0.13 | 4.19 | 1.51 | FFAS-3D | | ---------------EELEEAKRKLQARLAEAEETIESLNQKVIALEKTKQRLATEVEDLQLEVDRATAIANAAEGEWKLKVDDLAAELDASQKECRNYSTELFRLKGAYEEAQEQLEAVRRENKNLADEVKDLLDQIGEGGRNIHEIEKQKKRLEVEKDELQAAAEAALEQEENKVLRSQLELS---QVRQEIDRRIQEKEEEFENTRKNHQRALDS-MQASLEAEAKGKAEALRMKKKLEADINELEIALDHANKANSEAQKTIKKYQQQLKDVQTALERARDDAREQLGISERRANALQNELEESRTLLEQADRGRRQAEQELGDAHEQINELAAQ------------------------------------------------------------------------------- |
| 2 | 6yvuB | 0.08 | 0.08 | 3.26 | 1.00 | MapAlign | | LSLLEETQAKLKKNVETLEEKILAKKTHKQELQDLILDLKKKLNSLKDERSQGEKNFTSAHLKLKEMQKVLNAHRQRAMEARSSLSKAQNKSKVLTALSRLQVDKIERELSERENNFRVASDTVHEMEEELKKLRDHEPDLESQISKAEMEADSLASELTLAEQQVKEAEMAYVKAVSDKAQLNVVMKNLERLRGEYNDLQSETKTKKEKESVCQKLDILVAKLKKVKSASKKSGGDVVKFQKLLQNSERDVELSSDELKVIEEQLKHTKLALAENDTNMNETLNLKVELKEQSEQLKEQMEDMEESINEFKSIEIEMKNKLEKLNSLLTYIKSEILKKKRFDEFMAGFNIISMTLKEMYQMITMGGNAELELVDSLDPFSEGVTFSVMPPKKSWRNITNLSGGEKTLSSLALVFALH |
| 3 | 2tmaA | 0.14 | 0.09 | 3.19 | 1.14 | CNFpred | | ---------------------MDAIKKKMQMLKLDKENALDRAEQAEADKKAAEDRS------------------KQLEDELVSLQKKLKGTEDELDKYSEALKDAQEKLELAEKKATDAEADVASLNRRIQLVEEELDRAQERLATALQKLEEAEKAADESERGMKVIESRAQ------------------KDEEKMEIQEIQLKEHIAEDADRKYEEVARKLVIIESDLERAEERAELSEGKCAELEEEIKTVTNNLKSLEAQAEKYSQKEDKYEE----EIKVLSDKLKEAETRAEFAERSVTKLEKSIDDLEDELYAQKLKYKAISEELDHALNDMTSI--------------------------------------------------------------------------- |
| 4 | 3rj1B | 0.16 | 0.10 | 3.40 | 2.60 | HHsearch | | -----------------------------------------------------------------------------------------------------------------------------------------QEQFVKRRRD-LEHINLA-NESSLALEFVSLLLSSVKESTGSSSPFLTKKEYIELDLNESKDLLRASFNKLSSILQNEHDYWNKIQSKDIRKQIQLLKKII--FEKELYQIKKECALLISYGVSIENEFEIELLSLDDDEQDLPKINDIFKKTLRSRISRFANYKLLLKKIIKDYVLDIVPGSSINKEIRAFDKLLNIPRREGG--------------GGGGGGGGGGGGGGGGGGG-GGGGGGGGGGGGGGGGGGGGGGGGEVEDFLHFIVAEYIQQKK- |
| 5 | 6yvuB | 0.12 | 0.12 | 4.15 | 1.06 | SPARKS-K | | FEIVDREKNSLESGKETALEFLEKEKQLTLLRSKLFQFKLLQSNSKLASTLEKISSSNKDLEDEKMKFQESLKKVDEIKAQRKEIKDRISSCSSKEKTLVLERRELEGTRVSLEERTKNLVSKMEKAEKTLKSTKHSISEAENMLEELRGQQTEHETEIKDLTQLLEKERSILDDIKLSL-------KDKTKNISAEIIRHEKELEPWQLQEKESQIQLAESELSLLEETQAKLKKNVETLEEKILAKKTHKQELQDLILDLKKKLNSLKDERSQGEKNFTSAHLKLKEMQKVLNAHRQRAMEARSSLSKAQNKSKVLTALSRLQKSSFDVAISTACPRLLGYARFILLDRLRQFNLQPISTPENVPRLYSVLRDNVAYGKKRFRVVTVDGKLIDISGNHVAKGLMKLKVDDYTPEEV |
| 6 | 6f1tX | 0.24 | 0.16 | 4.99 | 1.91 | HHsearch | | --------------------------------------------VIRQ-----------------------------KEKDL---------------VLAARGKALLERNQDMSRQYEQMHKELTDKLEHLEQEKHELRRRFENRSELETDVKQLQDELERQQLHLREADREKTRAVQE----LSEQNQRLLDQLSRASEVERQLSMQVHALKE--------DFREKNSSTNQHIIRLESLQAEIKMLSDRKRELEHRLSATLEENDLLQGTVEELQDRVLI---------------LERQGHDKDLQLHQSQELQEVRLS-------YRQLQGGGGGGGGGGGGGGGG-GGGGGGGG-GGGGGGGGGGGGGGGGGGGG-GGGG--GGGGGGGGGGGGGGGG---------------- |
| 7 | 5j1iA | 0.12 | 0.08 | 2.81 | 1.12 | CNFpred | | ----------------------QRCISELKDIRLQLEACETRTVHR-EPARECAQRIAEQQ---------------KAQAEVEGLGKGVARLSAEAEKVL-AAPTLRSELELTLGKLEQVRSLSAIYLEKLKTISLVIRGTQGAEEVLRAHEEQLKEAQ-PELEATKASLKKLRAQAEAQQPTFD-------ALRDELRGAQEVGERLQQRHG-VEVERWRERVAQLLERWQAVLAQTDVRQRELEQLGRQLRYYRESADPLGAWLQDARRRQEQIQAM-SQAVREQLRQEQALLEEIERHGEKVEECQRFAKQYINAIKDYELQLVTYKAQLE------------------------------------------------------------------------------------ |
| 8 | 2tmaA | 0.13 | 0.09 | 3.07 | 1.49 | FFAS-3D | | ----------------------------MDAIKKKMQMLKLDKENALDRAEQAEADKKAAEDR-----------SKQLEDELVSLQKKLKGTEDELDKYSEALKDAQEKLELAEKKATDAEADVASLNRRIQLVEEELDRAQERLATALQKLEEAEKAADESERGMKVIESRAQK------------------DEEKMEIQEIQLKEAKHIAED-----ADRKYEEVARKLVIIESDLERAEERAELSEGKCAELEEEIKTVTNNLKSLEAQAEKYSQ----KEDKYEEEIKVLSDKLKEAETRAEFAERSVTKLEKSIDDLEDELYAQKLKYKAISEELDHALNDMTSI-------------------------------------------------------------------- |
| 9 | 6tpiA | 0.11 | 0.10 | 3.44 | 0.56 | CEthreader | | ----------------------------LKSIQADIAAKERAVRQKQQQRASLLAQLKKQEEA-----------ISEATRKLRETQNTLNQLNKQIDEMNASIAKLEQQKAAQERSLAAQLDAAFRQG-------EHTGIQLILSGEESQRGQRLQAYFGYLNQARQETIAQLKQTREE------------VAMQRAELEEKQSEQQTLLYEQRAQQAKLTQALNERKKTLAGLESSIQQGQQQLSELRANESRLRNSIARAEAAAKARAEREAREAQAVRDRQKEATRKGTTYKPTESEKSLMSRTGGLGAPRGQAFWPVRGPTLHRYGEQLQGELRWKGMVIGASEGTEVKAIADGRVILADWLQGYGLVVVVEHGKGDMSLYGYNQSALVSVGSQVRAGQPIALVGSSGGQGRPS |
| 10 | 5lm1A | 0.10 | 0.08 | 2.89 | 1.05 | SPARKS-K | | ------AHEASSLYSEEKAKLLREMMAKIEDKNEVLDQFMDSMDNLDAYSPQLMEKCAALSVR--------PDTVRNLVQSMQVLSGVFTDVEASLKDIRDLLEEDELLEQKFQEAVITSKAELAEVRREWAKYMEVHEKASFTNSELHRAMNLHVGNLRLLSG----PLDQVRAALPTPALSPEDKAVLQNRILAKVQEMRDQRVSLEQQLRELIQKDDITLFEEQLKKYDQLKVYLEQNLAAQDRVLCALTEANVQYAAVRRVLSDLDQKWNSTLQTLVASYEAYEDLMKKSQEGRDFYADLESKVAALLERTQSTCQAREAARQQLLDRE------------------------------------------------------------------------------------- |
| (a) | ID1 is the number of template residues identical to query divided by number of aligned residues. |
| (b) | ID2 is the number of template residues identical to query divided by query sequence length. |
| (c) | Cov is equal the number of aligned template residues divided by query sequence length. |
| (d) | Norm. Zscore is the normalized Z-score of the threading alignments. A Normalized Z-score >1 means a good alignment and is highlighted in bold. |
| (e) | Download alignment lists the threading program used to identify the template, and provide the 3D structure of aligned regions of threading templates (threading[1-10].pdb.gz). |
| (f) | Template residues identical to query sequence are highlighted in color. |
|