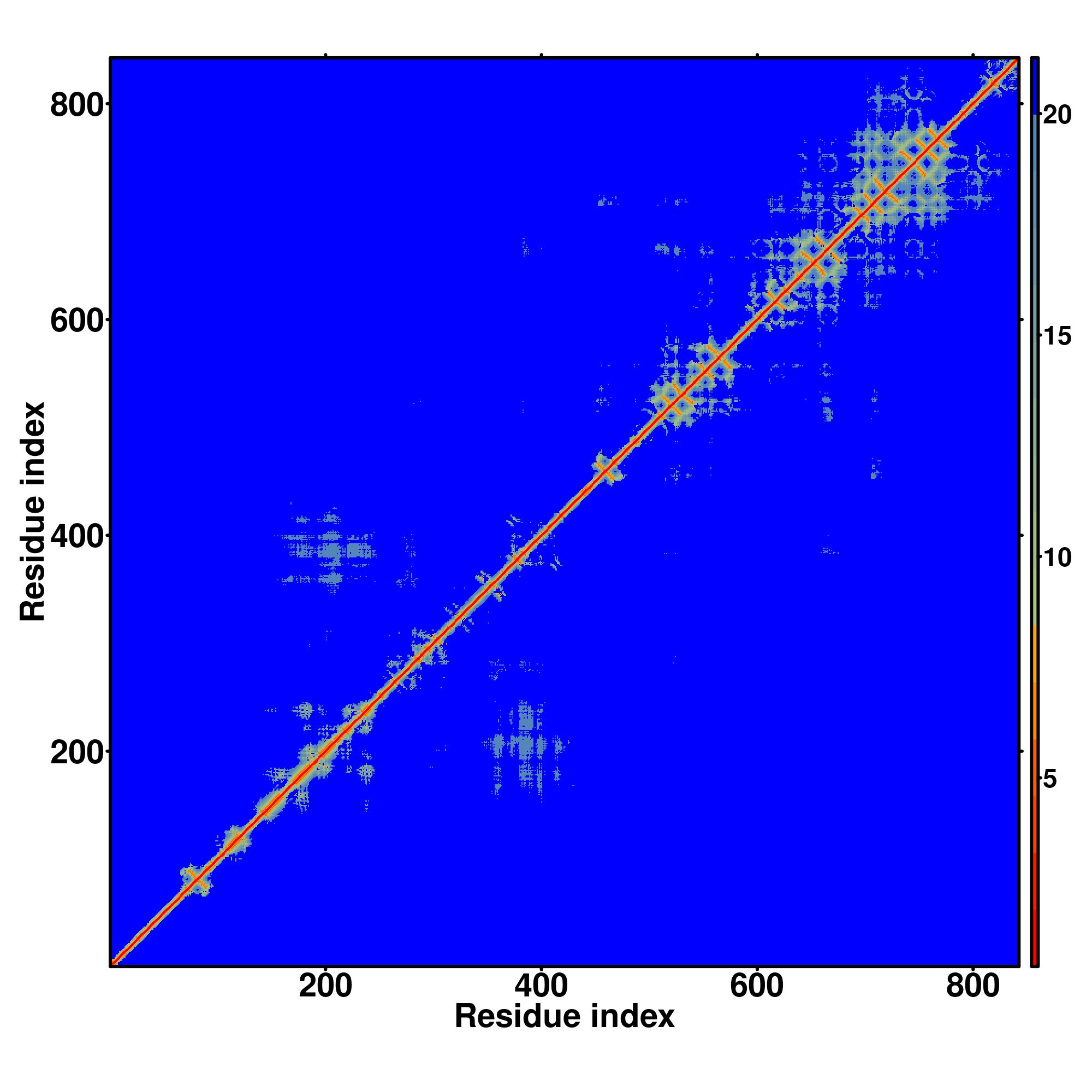
| Rank | PDB
hit | ID1 | ID2 | Cov | Norm.
Zscore | Downloadalignment | | 20 40 60 80 100 120 140 160 180 200
| | | | | | | | | | |
|---|
| SS
Seq | CCCCHHHHHHHHHHHHHCCCCCCCSSSSSSCCCSSSSCCCCCSSSSSSCCCCCCCCCHHHCCCCCCCCCCCSSSCCCSSSSCCSSSSSSCCCCCSSSSSSCCCCSSSSSSSCCCCCSSSSSCCCCCSSSSSSCCCCCCCCSSSSSSHHHCCCCCCCSSSCCCCCCCCCCCCCCCCCCCCSSSSSSSSCCCCCSSSSSHHHHSSSSSCC
FIEDSARKTLANILWREEGLSVGNMFYVFSDDGIIVIHPVDCEIQRHLKPTEKIFMSYEEICPQREKNATQPCQWVSAVNVRNRYIYVAQPALSRVLVVDIQAQKVLQSIGVDPLPAKLSYDKSHDQVWVLSWGDVHKSRPSLQVITEASTGQSQHLIRTPFAGVDDFFIPPTNLIINHIRFGFIFNKSDPAVHKVDLETMMPLKTIG |
|---|
| 1 | 5f30A | 0.07 | 0.06 | 2.62 | 1.33 | DEthreader | | ---CPIMHHMVVNYRIKYDITPEDGYAVGDKDICAEFDR-ETDMVRYAWA--F-DWDPNVRAWLDEDGKPSVANDALVFDPRGKWAVASMRLPGVCVVFDRENQVPVAVLAGPSAGHQAGFSPDGQSFLFMNSLR----QNNIMVWDSNPTTWEKKAVVESPDWRGYPNT-F-HMVFTPAKKIYVTMWWPNGIAVIDAVNWEVLKEVD |
| 2 | 6tv2A2 | 0.11 | 0.10 | 3.47 | 1.32 | SPARKS-K | | ----------------PPLRGSGDLGVLIERASVQILDGTAKTSLARVEGLGDLSHASLVFSRDQRYFGRDGGLTKLDLLAASDQAFVPAVGHHQVLVLDARDWKQTDAIDVAGQPVFVMTRPDDRQIWVNFAYPDN---DKVQVIDS-ETHEVIETLRP-------GPGVLHMEFSGRGDQVWISVRDADQLQVWDPYRLKRIGSLP |
| 3 | 3bwsA3 | 0.12 | 0.07 | 2.57 | 1.36 | FFAS-3D | | ---------------------------------------------------------------------TGKWSKILLYDPIRDLVYCSNWISEDISVIDRKTKLEIRKTDKIGLPRGLLLSKDGKELYIAQFSASNQESGGGRLGIYSDKEKLIDTIGPPGN-------KRHIVSGNTENKIYVSD-CCSKIEVYDLKEKKVQKSI- |
| 4 | 1l0qA | 0.15 | 0.12 | 3.90 | 2.22 | CNFpred | | --------NPMGAVISP----DGTKVYVANANDVSIIDTATNNVIATVPAG--------------------SSPQGVAVSPDGKQVYVTNMASSTLSVIDTTSNTVAGTVKTGKSPLGLALSPDGKKLYVTNNG-----DKTVSVINTVT-KAVINTVSVGR-------SPKGIAVTPDGTKVYVANFDSMSISVIDTVTNSVIDTVK |
| 5 | 3elqB2 | 0.09 | 0.09 | 3.44 | 1.33 | DEthreader | | GALPFGYLMGTAVWYEFDLETPNGTVLLRVRDHILEVDK-SGRVVDVWDLTKILDPKRDALLGAQAKLELGNHVNSIAYDAKDDSIILSSRHQ-GVVKIG-RDKQVKWILAFTYTQHTAWIS-SKGTLTIFDNGDQPMKYSRFVEYKIDEGTVQQVWEYGKERGDFYSPITSI-IEYQADNTMFGFGSIVGKLNEID-YKTKEVKEID |
| 6 | 3vgzA | 0.12 | 0.11 | 3.88 | 1.29 | SPARKS-K | | TQAIHNDLKPFGATINNTTQ----TLWFGNTVAVTAIDAKTGEVKGRLVLDDR----------KRTEEVRPLQPRELVADDATNTVYISGIGKSVIWVVDGGNIKLKTAIQNTGKSTGLALDSEGKRLYTTNAD------GELITIDT---ADNKILSRKKLLDDGKEHFFINISLDTARQRAFITDSKAAEVLVVDTRNGNILAKVA |
| 7 | 1fwxA1 | 0.10 | 0.09 | 3.18 | 0.50 | MapAlign | | -------DLHHVHMSFTEGKYDGRFLFMNDNTRVARVRCDVMKCDAILEI------------------PNAKGIHGLRPQKWSNYVFCNGEDENVFTAVDADKWEVAWQVLVSGNLDNCDADYEGKWAFSTSYSEKGAEMDHIVVFNIAEISLFTRYIPIAN------NPHGC-NMAPDKKHLCVAGKLSPTVTVLDVTRFAVVAEPE |
| 8 | 1l0qA | 0.17 | 0.12 | 3.97 | 0.39 | CEthreader | | ----------------------STFAYIANSDNISVIDVTSNKVTATIPVGSN---------------------PGAVISPDGTKVYVANAHSNDVSIIDTATNNVIATVPAGSSPQGVAVSPDGKQVYVTNAS------STLSVIDTTSNTVAGTVKTG--------KSPLGLALSPDGKKLYVTNNGDKTVSVINTVTKAVINTVS |
| 9 | 3vgzA | 0.13 | 0.12 | 4.02 | 0.99 | MUSTER | | RLDPVTLEVTQAIHNDLKPFGATQTLWFGNNSAVTAIDAKTGEVKGRLVLDDRKRTEEVRPL----------QPRELVADDATNTVYISGIGKSVIWVVDGGNIKLKTAIQNTGKSTGLALDSEGKRLYTTNAD------GELITIDTADNK---ILSRKKLLDDGKEHFFINISLDTARQRAFITDSKAAEVLVVDTRNGNILAKVA |
| 10 | 3u4yA1 | 0.09 | 0.07 | 2.56 | 0.72 | HHsearch | | --------------------TTSNFGIVVHLRRISFFSTDTLEILNQITLGY--------------------DFVDTAITSDCSNVVVTSDFCQTLVQIETQLEPKVVAIQEGQSSADVDITPDDQFAVTVTGLNHPFNQ-SYSFISTIPILIDPRP-----------LFGANQALNKTETKLFISANISRELKVFTIS-GKVVGYVA |
| (a) | ID1 is the number of template residues identical to query divided by number of aligned residues. |
| (b) | ID2 is the number of template residues identical to query divided by query sequence length. |
| (c) | Cov is equal the number of aligned template residues divided by query sequence length. |
| (d) | Norm. Zscore is the normalized Z-score of the threading alignments. A Normalized Z-score >1 means a good alignment and is highlighted in bold. |
| (e) | Download alignment lists the threading program used to identify the template, and provide the 3D structure of aligned regions of threading templates (threading[1-10].pdb.gz). |
| (f) | Template residues identical to query sequence are highlighted in color. |
|