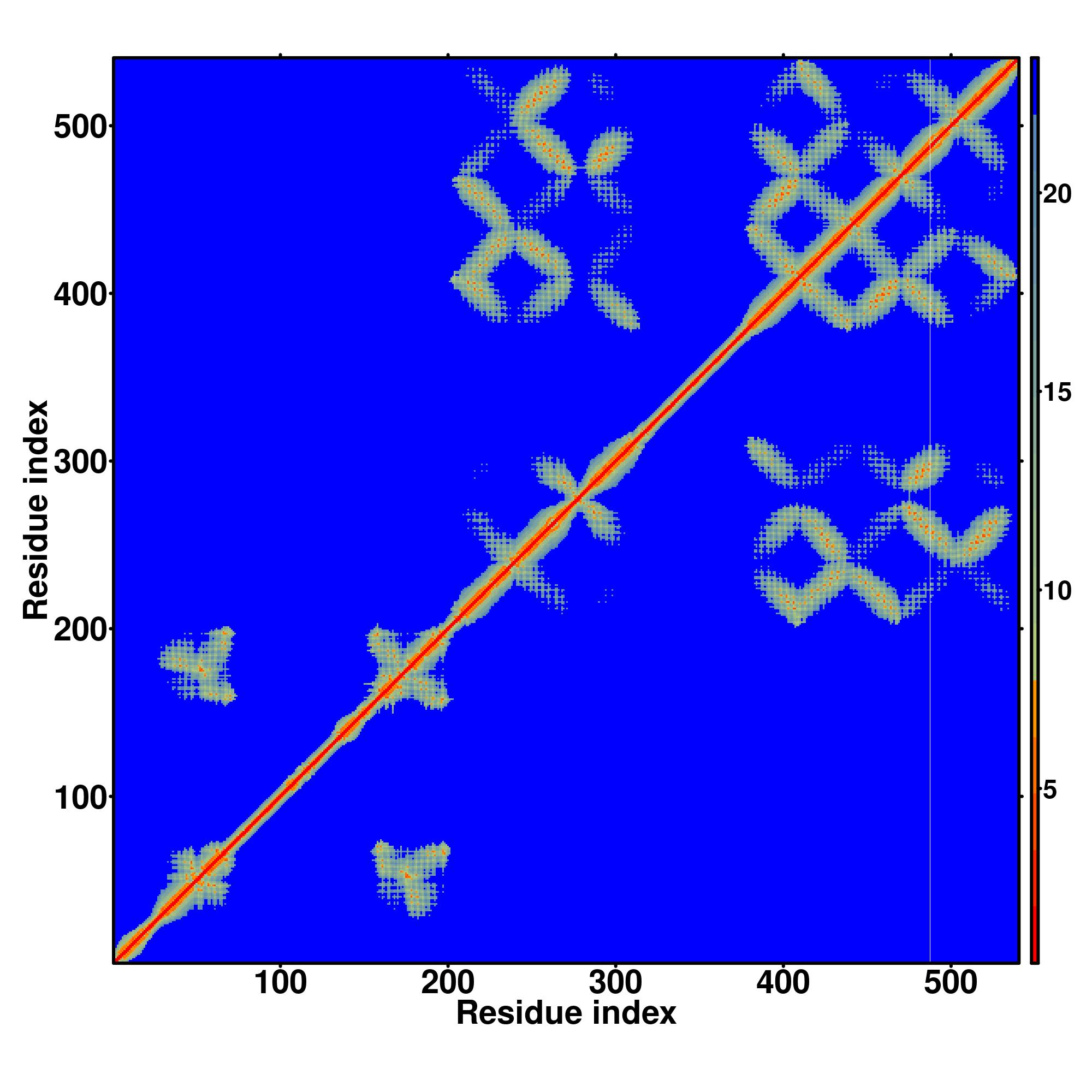
| Rank | PDB
hit | ID1 | ID2 | Cov | Norm.
Zscore | Downloadalignment | | 20 40 60 80 100 120 140 160 180 200 220 240 260
| | | | | | | | | | | | | |
|---|
| SS
Seq | CCCCCHHHHHHHHHHHHHHHHHHHHHHHHHHHHCCCHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHCCCCCCCCCHHHHHHHHHHHHHHHHHHHHHCCCCCCCCCHHHHHHHHHHHHHHHHHCCHHHHHHHHHHCCHHHHHHHHHHHHHHHCHHHHHHHHHHHHHCCCHHHHHHHHHHHHHHHHHHHHHHHHHHHCHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHCCCCCCHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHCCCC
APPGDLLSALLQSALAVLLLSLPSPLSLLLLRLLGPRLLRPLLGFLGALAVGTLCGDALLHLLPHAQEGRHAGPGGLPEKDLGPGLSVLGGLFLLFVLENMLGLLRHRGGTDITWMVLLGDGLHNLTDGLAIGAAFSDGFSSGLSTTLAVFCHELPHELGDFAMLLQSGLSFRRLLLLSLVSGALGLGGAVLGVGLSLGPVPLTPWVFGVTAGVFLYVALVDMLPALLRPPEPLPTPHVLLQGLGLLLGGGLMLAITLLEERLLPVTTEG |
|---|
| 1 | 5tsaA | 0.20 | 0.17 | 5.46 | 1.17 | DEthreader | | --N--R-VHVGYAVLGGAAGFAATALGALMALGLRA-ISARTQDAMLGFAAGMMLAASAFSLILPGLDAAGTIVGPGPAAAAVVALGLGLGVLLMLGLDYFT-----VNRVWLFVLTIILHNLPEGMAIGVSFA--TGDLRIGLPLTSAIAIQDVPEGLAVALALRAVGLPIGRAVLVAVASGLMEPLGALVGVGISSGFALAYPISMGLAAGAMIFVVSH-EV-------------HETTATVGLMAGFALMMFLDTAL---------- |
| 2 | 5tsaA | 0.24 | 0.20 | 6.36 | 2.21 | SPARKS-K | | -----NRVHVGYAVLGGAAGFAATALGALMALGLR-AISARTQDAMLGFAAGMMLAASAFSLILPGLDAAGTIVGPGPAAAAVVALGLGLGVLLMLGLDYFT--------VNRVWLFVLTIILHNLPEGMAIGVSFATGDRIGLPLTSAIAIQDVPEGLAVALALRAVGLPIGRAVLVAVASGLMEPLGALVGVGISSGFALAYPISMGLAAGAMIFVVSH--------EVHE------TTATVGLMAGFALMMFLDTAL---------- |
| 3 | 5tsaA | 0.24 | 0.20 | 6.35 | 1.50 | MapAlign | | -------VHVGYAVLGGAAGFAATALGALMALGLRA-ISARTQDAMLGFAAGMMLAASAFSLILPGLDAAGTIVGPGPAAAAVVALGLGLGVLLMLGLDYFT--------VNRVWLFVLTIILHNLPEGMAIGVSFATDLRIGLPLTSAIAIQDVPEGLAVALALRAVGLPIGRAVLVAVASGLMEPLGALVGVGISSGFALAYPISMGLAAGAMIFVVSH-E---V----------HETTATVGLMAGFALMMFLDTAL---------- |
| 4 | 5tsaA | 0.25 | 0.21 | 6.56 | 1.13 | CEthreader | | -NRVHVGYAVLGGAAGFAATALGALMALGLRA-----ISARTQDAMLGFAAGMMLAASAFSLILPGLDAAGTIVGPGPAAAAVVALGLGLGVLLMLGLDYFT--------VNRVWLFVLTIILHNLPEGMAIGVSFAGDLRIGLPLTSAIAIQDVPEGLAVALALRAVGLPIGRAVLVAVASGLMEPLGALVGVGISSGFALAYPISMGLAAGAMIFVVSHEV--------------HETTATVGLMAGFALMMFLDTAL---------- |
| 5 | 5tsaA | 0.25 | 0.21 | 6.66 | 1.49 | MUSTER | | -NRVHVGYAVLGGAAGFAATALGALMALGL------AISARTQDAMLGFAAGMMLAASAFSLILPGLDAAGTIVGPGPAAAAVVALGLGLGVLLMLGLDYF--------TVNRVWLFVLTIILHNLPEGMAIGVSFATGLRIGLPLTSAIAIQDVPEGLAVALALRAVGLPIGRAVLVAVASGLMEPLGALVGVGISSGFALAYPISMGLAAGAMIFVVSHEV--------------HETTATVGLMAGFALMMFLDTAL---------- |
| 6 | 5tsaA | 0.25 | 0.21 | 6.66 | 5.86 | HHsearch | | -NRVHVGYAVLGGAAGFAATALGALMALGL-----RAISARTQDAMLGFAAGMMLAASAFSLILPGLDAAGTIVGPGPAAAAVVALGLGLGVLLMLGLDYFTV--------NRVWLFVLTIILHNLPEGMAIGVSFATGLRIGLPLTSAIAIQDVPEGLAVALALRAVGLPIGRAVLVAVASGLMEPLGALVGVGISSGFALAYPISMGLAAGAMIFVVSHEV-------HE-------TTATVGLMAGFALMMFLDTAL---------- |
| 7 | 5tsaA | 0.25 | 0.21 | 6.55 | 2.17 | FFAS-3D | | --RVHVGYAVLGGAAGFAATALGALMALGL-----RAISARTQDAMLGFAAGMMLAASAFSLILPGLDAAGTIVGPGPAAAAVVALGLGLGVLLMLGLDYF--------TVNRVWLFVLTIILHNLPEGMAIGVSFATGLRIGLPLTSAIAIQDVPEGLAVALALRAVGLPIGRAVLVAVASGLMEPLGALVGVGISSGFALAYPISMGLAAGAMIFVVSHEVHETT--------------ATVGLMAGFALMMFLDT------------ |
| 8 | 5tsaA | 0.24 | 0.21 | 6.46 | 1.68 | EigenThreader | | ----NR-VHVGYAVLGGAAGFAATALGALMALGLRAIS-ARTQDAMLGFAAGMMLAASAFSLILPGLDAAGTIVGPGPAAAAVVALGLGLGVLLMLGLDYFT--------VNRVWLFVLTIILHNLPEGMAIGVSFATGLRIGLPLTSAIAIQDVPEGLAVALALRAVGLPIGRAVLVAVASGLMEPLGALVGVGISSGFALAYPISMGLAAGAMIFVVSH--------------EVHETTATVGLMAGFALMMFLDTAL---------- |
| 9 | 5tsaA | 0.25 | 0.21 | 6.44 | 1.38 | CNFpred | | ---------VGYAVLGGAAGFAATALGALMALGLR-AISARTQDAMLGFAAGMMLAASAFSLILPGLDAAGTIVGPGPAAAAVVALGLGLGVLLMLGLDYFT--------VNRVWLFVLTIILHNLPEGMAIGVSFATGLRIGLPLTSAIAIQDVPEGLAVALALRAVGLPIGRAVLVAVASGLMEPLGALVGVGISSGFALAYPISMGLAAGAMIFVVSHEV--------------HETTATVGLMAGFALMMFLDTAL---------- |
| 10 | 5oc9A | 0.06 | 0.05 | 2.23 | 1.00 | DEthreader | | MDSEALKKLETWYKYQLEYFTFALIPMAVIGLPYKYVIFASFNLIWSTVILELWKRGCANTTLLMKRKFEEPRPGKEPNHRLESAYQNHLILKVLVFNFLNCFASLYIALRSLATLLISQ-NQ--E-----------------------Y---L--LQFGYVSLFSCVYP-L-AAAFAVLNNFTEVNSDALKMCRFKRPFS--EP--SANIGVWQLAFET-MSVISVVTCAGVAVHALLALKFILAFA--IPDKPRHIQMKL--L----- |
| (a) | ID1 is the number of template residues identical to query divided by number of aligned residues. |
| (b) | ID2 is the number of template residues identical to query divided by query sequence length. |
| (c) | Cov is equal the number of aligned template residues divided by query sequence length. |
| (d) | Norm. Zscore is the normalized Z-score of the threading alignments. A Normalized Z-score >1 means a good alignment and is highlighted in bold. |
| (e) | Download alignment lists the threading program used to identify the template, and provide the 3D structure of aligned regions of threading templates (threading[1-10].pdb.gz). |
| (f) | Template residues identical to query sequence are highlighted in color. |
|