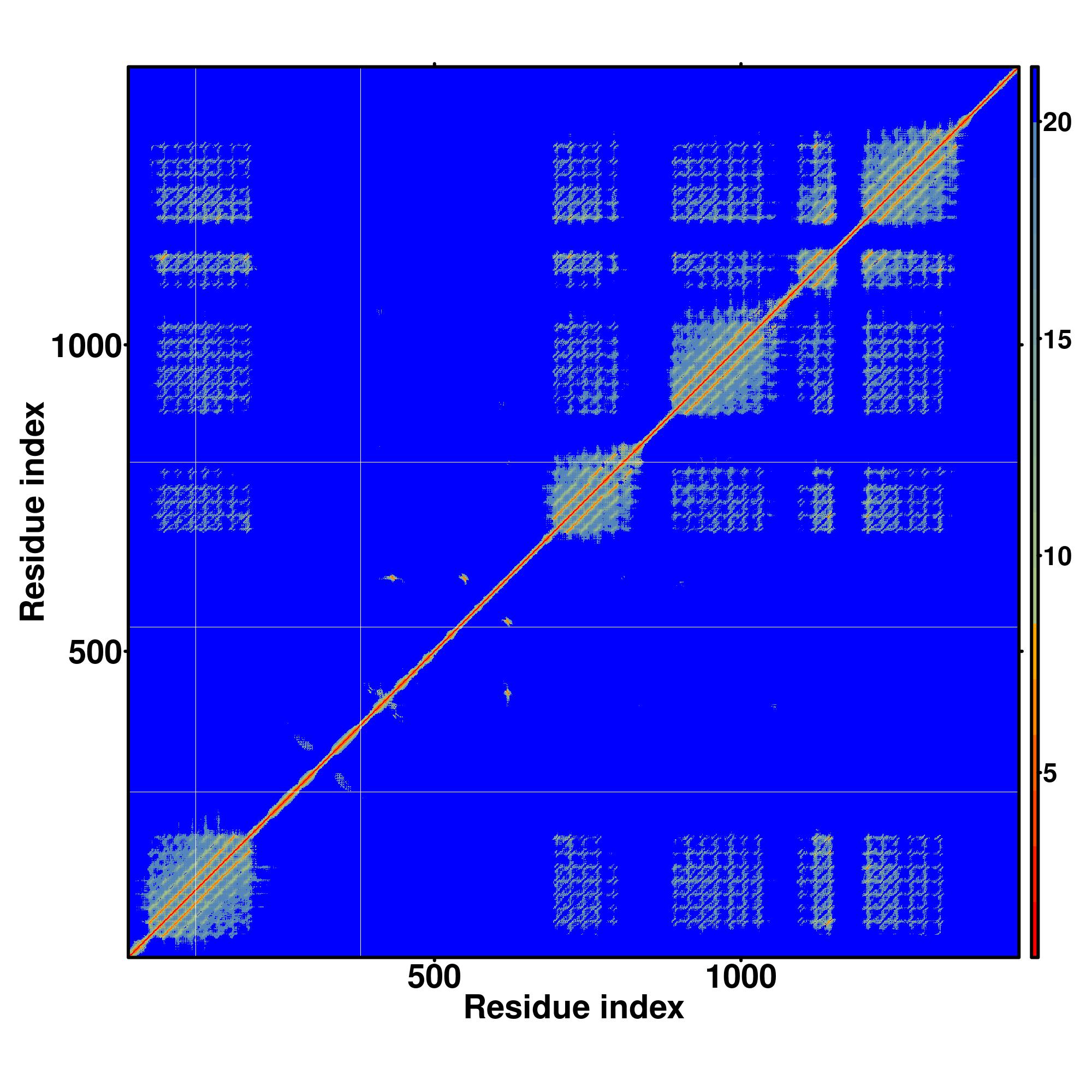
| Rank | PDB
hit | ID1 | ID2 | Cov | Norm.
Zscore | Downloadalignment | | 20 40 60 80 100 120 140 160 180 200 220 240 260 280 300 320 340
| | | | | | | | | | | | | | | | | |
|---|
| SS
Seq | CCCSSSSCCCCCCCCCCCCCCCCCCSSSCCCCCCCCSCCCCCCCCCCSSSCCCCCCCCCCCCCCCCCCCSSSCCCCCCCCCCCCCCCCCCCSSSCCCCCCCCCCCCCCCCCCCSSSCCCCCCCCCHHHHHCCCCCCSSSCCCCCCCCCHHHHHHCCCCCCCSSSCCCCCCCCCCCCCCCCHHHHHHHHCCCCCSCCCSSCCCCCCCCCHHHHHHHHHHHHHCCCCCCCCHHHHHHHHHHHHHHHHHHHCCCCCHHHHHHHHHHHHHHHHCCCCCHHHHHHHHHCCHHHHHHHHHHHHHHHCHHHHCCCHHHHCCCCCCCCCCCCCCCCCCCCCCCCCCCHHHHHSCCC
DTSKLEMFFLGYPRIVGLSLFPNLTSLTIVAQDIKEISGLEPCLQLKELWIAECCIEKIEGLQECRNLEKLYLYFNKISKIENLEKLIKLKVLWLNHNTIKNIEGLQTLKNLKDLNLAGNLINSIGRCLDSNEQLERLNLSGNQICSFKELTNLTRLPCLKDLCLNDPQYTTNPVCLLCNYSTHVLYHLPCLQRFDTLDVSAKQIKELADTTAMKKIMYYSHGLLIPLLLIELETVGNFHFEEGTRSDDWFNFCYELILSRFCAWDFRTYGITGVKVKRIIKVNNRILRLKFEEKFQKFLENEDMHDSESSKDLKFDDEVIKMEPRIKARPKLISLDDKTILSLAKTS |
|---|
| 1 | 4r5dA | 0.21 | 0.14 | 4.48 | 1.00 | DEthreader | | NLGWLNLSNNQLETLPVFEKLTNLTTLNLSNNQLTSLPVFERLASLTTLNLSNNQLTSLPVFERLTNLTTLNLSNNQLTSLPVFERLTNLTTLNLSNNQLTSLQVFERLTSLTTLNLSNNQLTSLPGVFERLTNLKTLNLSNNQLTKCRAVANAKQAASLHELHLSNNNIGEEGA-----A--ELVEALHPGSTLETLDLSNCNLTKACRERALKALELHLSNALVALLHPGS------------------------------A--TT-LHEPGST------------------------------------------------------------------------ |
| 2 | 4u06A | 0.19 | 0.17 | 5.38 | 2.19 | SPARKS-K | | EVRVLDLSRQELKTLPEIGKLKNLQRLYLHYNQLTVLPEIEQLKNLQLLYLRSNRLTTLPEIEQLKNLQVLDLGSNQLTVLQEIGQLKNLQLLYLHSNRLTTLKDIEQLQNLKSLDLSNNQLTTLPNEIEQLKNLKSLYLSENQFATF--PKEIGQLQNLKVLFLNNNQITILP------------NEIAKLKKLQYLYLSDNQLITLPKEIEQLQTLDLSYNQILPKEVGQLENLQTLDLR-NNQLKTLPKEIEQL-------KNLQTLFLSNNQLTILP------------QEIGKL---------KNLLWLSLVYNQLTTPNEIEQLKNLQTLYNNQFSSQEKKR |
| 3 | 4r5dA | 0.16 | 0.15 | 5.06 | 0.50 | MapAlign | | NVRYLALGGNKLHDISALKELTNLGWLNLSNNQLETLPVFEKLTNLTTLNLSNNQLTSLPVFERLASLTTLNLSNNQLTSLPVFERLTNLTTLNLSNNQLTSLPVFERLTNLTTLNLSNNQLTSLPGVFERLTSLTTLNLSNNQLTSL-PDGVFERLTNLKTLNLSNNQLTKE--------ACRAVANAKQAASLHELHLSNNNIGEEGAAELVETLDLSNCNLTKEACREIARALKTTLHELHLSNNNIGEEGAAELVEALLHPGS---TLETLDLSNC--NLTKEACREIARALLHELHLSNNNIGEEGAAELVEALLHPGSTLETLDLSNCNLTKEACREIA--- |
| 4 | 6mkyA | 0.26 | 0.19 | 5.86 | 0.31 | CEthreader | | -VKTLCLRQNLIKCIENLEELQSLRELDLYDNQIKKIENLEALTELEILDISFNLLRNIEGVDKLTRLKKLFLVNNKISKIENLSNLHQLQMLELGSNRIRAIENIDTLTNLESLFLGKNKITKLQ-NLDALTNLTVLSMQSNRLTKIE---GLQNLVNLRELYLSHNGIEVIE-------------GLENNNKLTMLDIASNRIKKIENISHLTELQEFWMNDNSWSDLDELKGARSLE-TVYLERNPLQKDPQYRRKVMLALPSVRQIDATFV------------------------------------------------------------------------- |
| 5 | 5il7A | 0.22 | 0.21 | 6.63 | 1.55 | MUSTER | | SLSELSLSSNQITDISPLASLNSLSMLWLDRNQITDIAPLASLNSLSMLWLFGNKISDIAPLESLKSLTELQLSSNQITDIAPLASLKSLTELSLSGNNISDIAPLESLKSLTELSLSSNQITDI-APLASLKSLTELSLSSNQISDI---APLESLKSLTELQLSRNQISDIPLESLKSLTETDIAPLASLKSLTELQLSRNQISDIAPLESLNSLSKLWLNGNQIAPLASLNSLTELELSSNQITD---------IAPLASLKSLSTLWLSSNQISDIAPLASLESLSELSLSSNQISDISPLASLNSLTGFDVRRNPIKRLPETITGFDMEILWNDGFITFFDNP |
| 6 | 6hluA | 0.25 | 0.23 | 7.03 | 0.81 | HHsearch | | SLSELSLSSNQITDISPLASLNSLSMLWLDRNQITDIAPLASLNSLSMLWLFGNKISDIAPLESLKSLTELQLSSNQITDIAPLASLKSLTELSLSGNNISDIAPLESLKSLTELSLSSNQITDIAP-LASLKSLTELSLSSNQISDIAP---LESLKSLTELQLSRNQISDIAP-------------LESLKSLTELQLSSNQITDIAP-LASKSLLQLSRNSDIAP-LESLNSLSKLWLNGNQITDIPLASL-----NSLTELELSSNQITDLKSLSTLWLS-----------SNQISDIAPLASLESLSELSLSSNQISDISPLASLNSLTGFDRNPIKRLPETI |
| 7 | 4r58A | 0.26 | 0.15 | 4.69 | 1.67 | FFAS-3D | | AFAETIKANLKKKSVTDQNELNSIDQIIANNSDIKSVQGIQYLPNLKTLKLSNNKITDISALKQLNNLGWLDLSNNGITDISALKNLASLHTLDLSNNGITDISALKNLDNLHTLDLSNNGITD-ISALKNLDNLHTLDLSNNGITDI---SALKNLTSLHTLDLSNNGITDI-------------SALKNLDNLETLDLRNNGITDKSALKNLNNLK---------------------------------------------------------------------------------------------------------------------------------- |
| 8 | 4r5dA | 0.17 | 0.16 | 5.37 | 0.87 | EigenThreader | | SIDQIIANNSDIKSVQGIQYLPNVRYLALGGNKLHDISALKELTNLGWLNLSQLETLPQGVFEKLTNLTTLNLSNNQLTSLGVFERLASLTTLNLSNNQLTSLPVFERLTNLTTLNLSNNQLTSLPQGVFRLTNLTTLNLSNNQLTSLPQGVFER-LTSLTTLNLSNNQLTSLP------DGV-----FERLTNLKTLNLSNNQLTKEACRAVANALKQANNNIGEEGAAELVEALLHPGSTSNCNLTKEACREIARALKQAAAELVEALLHPGSTLETLDLSLTKEACREIARALK------QATTLHELHLSNNNIGEEGAAELVEALLHPGSTLEREIARALKQA |
| 9 | 4r5dA | 0.20 | 0.18 | 5.60 | 5.51 | CNFpred | | SIDQIIANNSDIKSVQGIQYLPNVRYLALGGNKLHDISALKELTNLGWLNLSNNQLETLPVFEKLTNLTTLNLSNNQLTSLPVFERLASLTTLNLSNNQLTSLPVFERLTNLTTLNLSNNQLTSLPQVFERLTNLTTLNLSNNQLTSLPQG-VFERLTSLTTLNLSNNQLTSLPDGV-----------FERLTNLKTLNLSNNQLTKEACRA-------------VANALKQAASLHELHLS----NNNIGEEGAAELVEALLHP----------STLETLDLSNCNLTKEACREIARALK-----QATTLHELHLSNNNIGEE-GAAELVEALLHPGSTLETLDLSN |
| 10 | 4u08A | 0.20 | 0.14 | 4.32 | 1.00 | DEthreader | | NVRILNLSGSKLTTLPEIGKLQNLQLLNLDDNQLIALPEIGKLQNLQQLHLSKNQLALPEEIGQLQNLQKLKLYENQLTAIPEIGQLQNLQELNLAHNQLATLPDIEQLQRLQTLYLGHNQFNSILKEIGQLQNLESLGLDHNQLNVL-P-KEIGQLRNLESLGLDHNQL--N-V--------LP-KEIGQLQNLQILHLRNNQL-TTLPKEIGQLLKLLLNK-LLKEIGQL------------------PK-EI--GQ--L----Q--NLQELPENIGQ-------------------------------------------------------------------- |
| (a) | ID1 is the number of template residues identical to query divided by number of aligned residues. |
| (b) | ID2 is the number of template residues identical to query divided by query sequence length. |
| (c) | Cov is equal the number of aligned template residues divided by query sequence length. |
| (d) | Norm. Zscore is the normalized Z-score of the threading alignments. A Normalized Z-score >1 means a good alignment and is highlighted in bold. |
| (e) | Download alignment lists the threading program used to identify the template, and provide the 3D structure of aligned regions of threading templates (threading[1-10].pdb.gz). |
| (f) | Template residues identical to query sequence are highlighted in color. |
|