| Rank | PDB
hit | ID1 | ID2 | Cov | Norm.
Zscore | Downloadalignment | | 20 40 60 80 100 120 140 160 180 200 220 240 260 280 300 320 340 360 380 400 420
| | | | | | | | | | | | | | | | | | | | | |
|---|
| SS
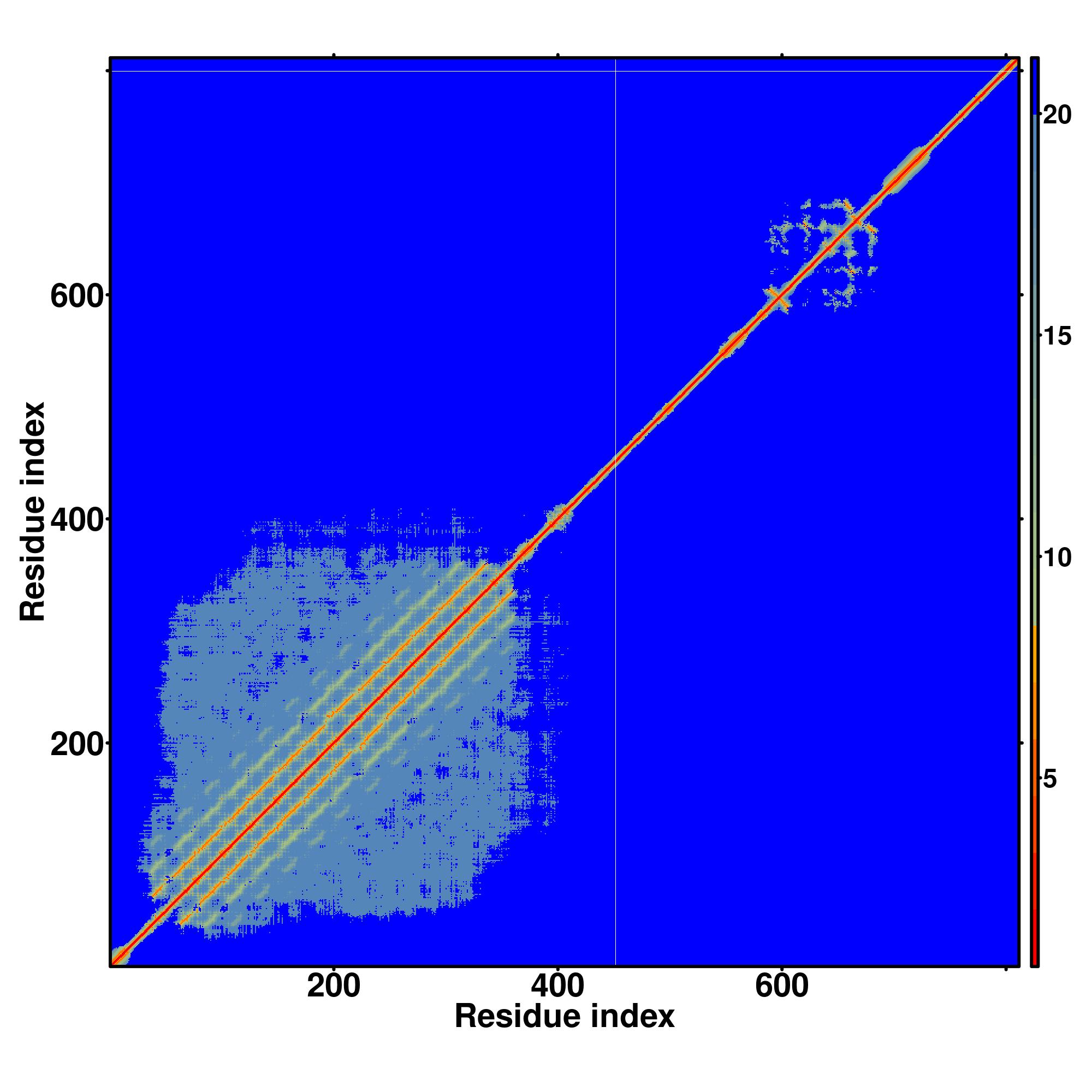
Seq | CCCHHHHHHHHHHHHHHHCCCCCCCCCCCCCSCCCCCSSSSCCCCCCSCCCCCCCCCCCCCCSSSCCCCCCCCCCHHHHCCCCCCCSSSCCCCCCCCCCHHHHCCCCCCCSSSCCCCCCCCCCHHHHCCCCCCCSSSCCCCCCCCCCHHHHCCCCCCCSSSCCCCCCCSCCHHHCCCCCCCCSSSCCCCCCCCCCHHHHHCCCCCCSSSCCCCCCCCCCCCHHHHCCCCCCCSSSCCCCCCCCCCCHHHCCCCCCCSSSCCCCCCCCCCHHHHCCCCCCCSSSCCCCCCCCCCHHHHCCCCCCCSSSCCCCCCCCCCHHHHCCCCCCCSSSCCCCCCCCCCHHHHCCCCCCCSSSCCCCCCCCCCCHHHHHHHHHHCCCCCCCCCCCCSCCCCHHHCCCSCCCCCHHHCCCCCCCCCCCCCCCCCCC
MEAARALRLLLVVCGCLALPPLAEPVCPERCDCQHPQHLLCTNRGLRVVPKTSSLPSPHDVLTYSLGGNFITNITAFDFHRLGQLRRLDLQYNQIRSLHPKTFEKLSRLEELYLGNNLLQALAPGTLAPLRKLRILYANGNEISRLSRGSFEGLESLVKLRLDGNALGALPDAVFAPLGNLLYLHLESNRIRFLGKNAFAQLGKLRFLNLSANELQPSLRHAATFAPLRSLSSLILSANNLQHLGPRIFQHLPRLGLLSLRGNQLTHLAPEAFWGLEALRELRLEGNRLSQLPTALLEPLHSLEALDLSGNELSALHPATFGHLGRLRELSLRNNALSALSGDIFAASPALYRLDLDGNGWTCDCRLRGLKRWMGDWHSQGRLLTVFVQCRHPPALRGKYLDYLDDQQLQNGSCADPSPSASLTADR |
|---|
| 1 | 4u7lA | 0.28 | 0.26 | 8.00 | 1.33 | DEthreader | | --------S----STRS-LPNLQEVYLNELVPLSHVVSLFLQH-NKIRSVEGSQLKAYLSLEVLDLSLNNITEVRNTCFPHGPPIKELNLAGNRIGTLELGAFDGLSSLLTLRLSKNRITQLPVRAF-KLPRLTQLDLNRNRIRLIEGLTFQGLNSLEVLKLQRNNISKLTDGAFWGLSKMHVLHLEYNSLVEVNSGSLYGLTALHQLHLSNNSIAR-I-HRKGWSFCQKLHELVLSFNNLTRLDEESLAELSSLSVLRLSHNSISHIAEGAFKGLRSLRVLDLDHNEISGTTSGAFSGLDSLSKLTLFGNKIKSVAKRAFSGLEGLEHLNLGGNAIRSVQFDAFVKMKNLKELHISSDSFLCDCQLKWLPPWLIGRM---LQAFVTATCAHPESLKGQSIFSVPPESFV-CDDF------------ |
| 2 | 4u7lA | 0.27 | 0.26 | 7.98 | 2.67 | SPARKS-K | | -CPSRCTCSGDSLDCGGRGLAALPGDLPSSTDLPNLQEVYLNNNELTAVPSL--GAASSHVVSLFLQHNKIRSVEGSQLKAYLSLEVLDLSLNNITEVRNTCFPHGPPIKELNLAGNRIGTLEPVRAFKLPRLTQLDLNRNRIRLIEGLTFQGLNSLEVLKLQRNNISKLTDGAFWGLSKMHVLHLEYNSLVEVNSGSLYGLTALHQLHLSNNSIARI--HRKGWSFCQKLHELVLSFNNLTRLDEESLAELSSLSVLRLSHNSISHIAEGAFKGLRSLRVLDLDHNEISGTTSGAFSGLDSLSKLTLFGNKIKSVAKRAFSGLEGLEHLNLGGNAIRSVQFDAFVKMKNLKELHISSDSFLCDCQLKWLPPWLIGRMLQAF---VTATCAHPESLKGQSIFSVPPESFVCDDFLKA---------- |
| 3 | 4u7lA | 0.28 | 0.26 | 7.96 | 0.68 | MapAlign | | -SLDCGGRGLAALPGDLPSSTRSLNLSYNKLSLPNLQEVYLNNNELTAV--PSLGAASSHVVSLFLQHNKIRSVEGSQLKAYLSLEVLDLSLNNITEVRNTCFPGP-PIKELNLAGNRITQLPVRAFK-LPRLTQLDLNRNRIRLIEGLTFQGLNSLEVLKLQRNNISKLTDGAFWGLSKMHVLHLEYNSLVEVNSGSLYGLTALHQLHLSNNSIA-RI-HRKGWSFCQKLHELVLSFNNLTRLDEESLAELSSLSVLRLSHNSISHIAEGAFKGLRSLRVLDLDHNEISIETSGAFSGLDSLSKLTLFGNKIKSVAKRAFSGLEGLEHLNLGGNAIRSVQFDAFVKMKNLKELHISSDSFLCDCQLKWLPPWLIGR----MLQFVTATCAHPESLKGQSIFSVPPESFVC---------------- |
| 4 | 4u7lA | 0.27 | 0.26 | 7.92 | 0.43 | CEthreader | | EIDPAGFEDLPNLQEVYLNNNELTAVPSLGAASSHVVSLFLQHNKIRSVE-GSQLKAYLSLEVLDLSLNNITEVRNTCFPHGPPIKELNLAGNRIGTLELGAFDGLSSLLTLRLSKNRITQLPVRAF-KLPRLTQLDLNRNRIRLIEGLTFQGLNSLEVLKLQRNNISKLTDGAFWGLSKMHVLHLEYNSLVEVNSGSLYGLTALHQLHLSNNSIARIH--RKGWSFCQKLHELVLSFNNLTRLDEESLAELSSLSVLRLSHNSISHIAEGAFKGLRSLRVLDLDHNEISGTTSGAFSGLDSLSKLTLFGNKIKSVAKRAFSGLEGLEHLNLGGNAIRSVQFDAFVKMKNLKELHISSDSFLCDCQLKWLPPWLIGRM---LQAFVTATCAHPESLKGQSIFSVPPESFVCDDFLKA---------- |
| 5 | 5a5cA | 0.34 | 0.26 | 7.92 | 2.04 | MUSTER | | ------------------------MACPPKCRCEK-LLFYCDSQGFHSVPNGL----PSQLLGLSLRHNQLQSLPNGVFDKLTQLTWLHLDHNQLQSLPNGVFDKLTKLTELILSSNQLQSLPNGTFDKLTNLQNLDLSFNQLQSLPNGVFDKLTNLQTLHLRSNQLQSLP--------------------------------------------------NGVFDKLTSLTFLDLSTNQLQSLPNGVFDKLTNLRELHLEHNQLQSLPNGVFDKLTSLTTLFLQWNQLQSLPNGVFDKLTNLEKLDLTGNQLQSLPNGVFDKLTNLKILLLDNNQLQSLPNGVFDKLKSLTTVGLSGNLWECSPRVCALASWLGSF---QGRWEHSILCHSPDHTQGEDILDAVHGFQLCW--------------- |
| 6 | 4u7lA | 0.29 | 0.26 | 7.91 | 1.01 | HHsearch | | --------------------------CPSRCTCSG-DSLDCGGRGLAALPGDL----PSSTRSLNLSYNKLSEIDPAGFEDLPNLQEVYLNNNELTAVEGSQLKAYLSLEVLDLSLNNITEVRNTCFPHGPPIKELNLAGNRIGTLELGAFDGLSSLLTLRLSKNRITQLPVRTFQGLNSLEVLKLQRNNISKLTDGAFWGLSKMHVLHLEYNSLVEVN--SGSLYGLTALHQLHLSNNSIARIHEESLAELSSLSVLRLSHNSISHIAEGAFKGLRSLRVLDLDHNEISGTISGAFSGLDSLSKLTLFGNKIKSVAKRAFSGLEGLEHLNLGGNAIRSVQFDAFVKMKNLKELHISSDSFLCDCQLKWLPPWLIGRML---QAFVTATCAHPESLKGQSIFSVPPESFVCDDFLKA---------- |
| 7 | 2id5B | 0.27 | 0.24 | 7.39 | 2.65 | FFAS-3D | | --------------------------CPPRCECAQDRAVLCHRKRFVAVPEGI----PTETRLLDLGKNRIKTLNQDEFASFPHLEELELNENIVSAVEPGAFNNLFNLRTLGLRSNRLKLIPLGVFTGLSNLTKLDISENKIVILLDYMFQDLYNLKSLEVGDNDLVYISHRAFSGLNSLEQLTLEKCNLTSIPTEALSHLHGLIVLRLRHLNINA--IRDYSFKRLYRLKVLEISHWPYLDTMTPNCLYGLNLTSLSITHCNLTAVPYLAVRHLVYLRFLNLSYNPISTIEGSMLHELLRLQEIQLVGGQLAVVEPYAFRGLNYLRVLNVSGNQLTTLEESVFHSVGNLETLILDSNPLACDCRLLWVFRRR----WRLNFNRQQPTCATPEFVQGKEFKDFPDVLLPNY--------------- |
| 8 | 4lxrA | 0.22 | 0.20 | 6.37 | 0.95 | EigenThreader | | DTTDYQQL-------------PKKLRICMLPGHTPPTTLISDNLGMN-ITRQHLDRLHGLKRFRTTRRLTHI--PANLLTDMRNLSHLEA---NIEEMPSHLFDDLENLESIEFGSNKLRQMPRGIFGKMPKLKQLNLWSNQLHNLTKHDFEGATSVLGIDIHDNGIEQLPHDVFAHLTNVTDINLSANLFRSLPQGLFDHNKHLNEVRLMNNRVPLATLPSRLFANQPELQILRLRAELQS-LPGDLFEHSTQITNISLGDNLLKTLPATLLEHQVNLLSLDLSNNRLTHLPDSLFAHTTNLTDLRLEDNLLTGISGDIFSNLGNLVTLVMSRNRLRTIDSRAFVSTNGLRHLHLDHNDIDLQQPLLDIMLQTQINSPFGYMHGLLTLNLRNNSIIFVYNDWKNT---------MLQLRELDLSYN |
| 9 | 4oqtA | 0.28 | 0.25 | 7.51 | 9.05 | CNFpred | | --------------------------CPPRCECSADRAVLCHRKRFVAVPE----GIPTETRLLDLGKNRIKTLNQDEFASFPHLEELELNENIVSAVEPGAFNNLFNLRTLGLRSNRLKLIPLGVFTGLSNLTKLDISENKIVILLDYMFQDLYNLKSLEVGDNDLVYISHRAFSGLNSLEQLTLEKCNLTSIPTEALSHLHGLIVLRLRHLNINAIR--DYSFKRLYRLKVLEISHWYLDTMTPNCLYGL-NLTSLSITHCNLTAVPYLAVRHLVYLRFLNLSYNPISTIEGSMLHELLRLQEIQLVGGQLAVVEPYAFRGLNYLRVLNVSGNQLTTLEESVFHSVGNLETLILDSNPLACDCRLLWVFRRRWRLNF-----RQQPTCATPEFVQGKEFKDFPDVLL------------------ |
| 10 | 2a0zA | 0.25 | 0.23 | 7.06 | 1.33 | DEthreader | | --------K--FGSIRN-LKWTNLLDLSNLVSFLQLEYFFLEYNNIQH-LFSHSLHGLFNVRYLNLKRSFTPKIDDFSFQWLKCLEHLNMEDNDIPGIKSNMFTGLINLKYLSLSNSFLRTLTNETFVAHSPLHILNLTKNKISKIESDAFSWLGHLEVLDLGLNEIGQLTGQEWRGLENIFEIYLSYNKYLQLTRNSFALVPSLQRLMLRRVALKNVDSSPSPFQPLRNLTILDLSNNNIANINDDMLEGLEKLEILDLQHNNLALWPIYFLKGLSHLHILNLESNGFDEIPVEVFKDLFELKIIDLGLNNLNTLPASVFNNQVSLKSLNLQKNLITSVEKKVFGPARNLTELDMRFNPFDCTCESI--AWFVNWINETHTNIPLHYLCNTPPHYHGFPVRLFDTSS-C----------------- |
| (a) | ID1 is the number of template residues identical to query divided by number of aligned residues. |
| (b) | ID2 is the number of template residues identical to query divided by query sequence length. |
| (c) | Cov is equal the number of aligned template residues divided by query sequence length. |
| (d) | Norm. Zscore is the normalized Z-score of the threading alignments. A Normalized Z-score >1 means a good alignment and is highlighted in bold. |
| (e) | Download alignment lists the threading program used to identify the template, and provide the 3D structure of aligned regions of threading templates (threading[1-10].pdb.gz). |
| (f) | Template residues identical to query sequence are highlighted in color. |
|