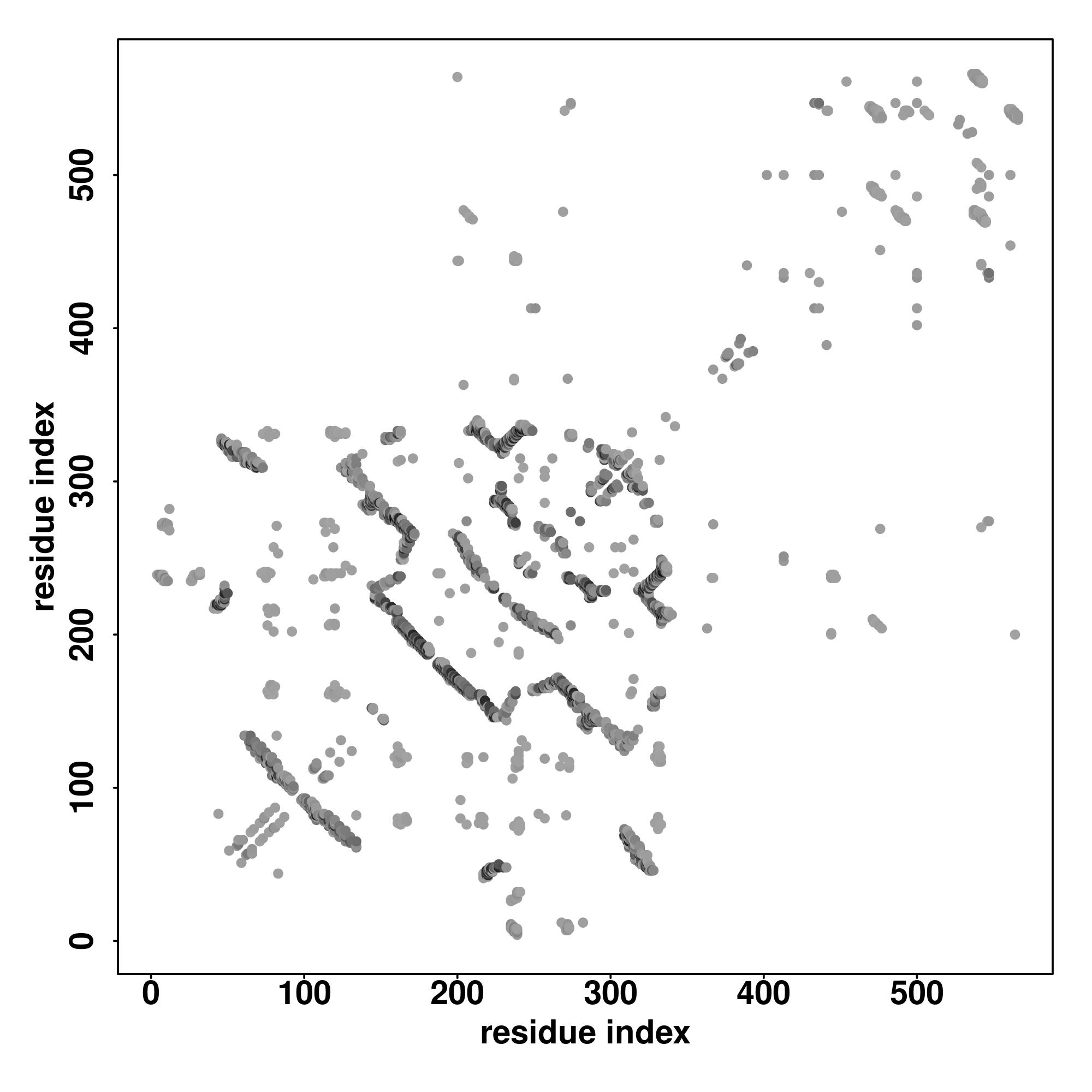
| Rank | PDB
hit | ID1 | ID2 | Cov | Norm.
Zscore | Downloadalignment | | 20 40 60 80 100 120 140 160 180
| | | | | | | | | |
|---|
| SS
Seq | CCCCSSSCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCHHHHHCCHHHHHHCCCCCCCCCCCCCSCCCCCCCCCCCCCCCCCCCCCSCCCCCCCCCCCCSSCCCCCCCCCCCCCCCHHHHHHHHHCCCCCCCCCCCCCCCCCCCCCCCCSSSSSCCCCCCCCCCCCSSCCCCCSSCCC
YSEAVLMDLAGLGTRCGGAGGGYAPSCRYGGVGGPGAAGVAPYRRSCEHCQRAVSSSSIFSRSALSICASPRAGPGPGGGAGCGGSRFSLGRLYGSRRSCLWRSRSGSVNEASCPTEQTRLSSQASMGDDEDDDEEEAGPPPPYHDALYFPVLIVHRQEGCLGHSHRPLHRHGSCVETSL |
|---|
| 1 | 5hq4A1 | 0.06 | 0.06 | 2.62 | 0.48 | CEthreader | | --ATVQSPDGNIKIIISDEQSTPSYSISFKNKTVINNSALGFEFKQHAPFSNSFKITKVQQQSTNTQWQQPW---GERQTVVDQHNEVTVTFAKGGTYSVRFKAFDSGVGFRYEVPKQAGLNNIEITKELTEFAVNNSHTATAWWIPARGWNRYEYVYNTTPLNDAALVHTPFTFKNQDG |
| 2 | 1jb0A2 | 0.07 | 0.07 | 2.95 | 0.55 | EigenThreader | | GGAAHGAIFMVRDYDTLAPGGTAPNAAATASVAFGGDVVAVGVHHIHAFTIHVTVLILLKGVLFAFRFPCDGPGRGGTCQVSGWDHVFLGLFWMYNCISVVIFHFSWKMQSDVRDFLWAQASQVIGSYGSALSAYGLLFLGAHFIWAFSLMFLFSLKVAPAIQPRALSIIQGRAVGVAHY |
| 3 | 2ftcC | 0.09 | 0.08 | 3.14 | 0.33 | FFAS-3D | | --KMATLSVGGKTVSRFRKATSILEFYRELGLPPKQTVKIFNITDNAAIKPGTPLYAAHFRPGQYVDVTAKTIGKGFQG--VMKRWGFKGQATHGQTKT---HRRPGAVA----TGDIGRVWPGTKMPGKMGNIYRTEYGLKVWRINTKHNIIYVNGSVPGHKNCLVKVKDS-------- |
| 4 | 7jjvA | 0.13 | 0.09 | 3.09 | 0.82 | SPARKS-K | | ----------MQCDGLDGADGTSNGQAGASGLAGGPNCNGGKGGKGAPGVGTAGGAGGVGGAGGTGNTNGGAGGSGGNSDVAAGGAGAAGGAAGGAGTGG--TGGNGGAGKPGGPGAGGAGTPAGSAGSPGQTTVL-------------------------------------------- |
| 5 | 1m3dC | 0.25 | 0.07 | 2.24 | 0.36 | CNFpred | | ----------------------------------------------------------------------------------------------------LLVKHSQTDQEPMCPVGMNKL-------------------------WSGYSLLYFEGQEKAHN---QDLGLAGSCLARFS |
| 6 | 2x0sA | 0.10 | 0.07 | 2.50 | 0.67 | DEthreader | | ---KKWVYYFGGGNAD-----------NLAEMVPVPPGFTI----------TETIPQVADQVRENVSRVEK------EMGF-FSVRS----------------NLGLSYRRITMYAMQVGREDEEALSRMLDGYLELFESFPQDPVMQLFAAIKAFRSWGNPRAIYRGTAVNV------- |
| 7 | 5necA | 0.11 | 0.11 | 3.84 | 0.76 | MapAlign | | -REDSKRDGYIVDTNTGLGSNKCNPSLIGAPSGYNCTSNPHDPWNGSITRKYAPLNTVGTTKAIYAFDTIDLNEQWQVNIGARFDSFETTAKNHRPATKLSDKSSGSIYASYATSAETTNYELGTKWAFFNERLELSAAIFRTDKDNTQSRVDGVELSASGKLTEKWKVFAGYSYLDSEL |
| 8 | 4k0mC | 0.08 | 0.08 | 3.08 | 0.62 | MUSTER | | FDETV--EVHAKLGIDPRRSDQNVRGTVSLPHGLGKQVRVLAIAKG-EKIKEAEEAGADYVGGEEIIQAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA |
| 9 | 3iv0A3 | 0.09 | 0.06 | 2.30 | 0.52 | HHsearch | | ----SLLDI----------------------------NRSSVIFAG-------GSDEGLFEIAQNINFNE-------IF-----NA--KFSDNVSYSKSPLFCYSGDYL-TL-FPYDDARKELWFDE----KIYSTSVSSSAPKEIKKFWNIDT-YGNTITSNSGNQIVFR---YAGALL |
| 10 | 4epaA | 0.07 | 0.07 | 2.95 | 0.46 | CEthreader | | TRASIGNVKLRLAPDDQPWEGFAASRECTRATQDAYVGWNDIKGRKLSISDGSPDPYRRCTDSQTLSGKYTTDDWVFNLISAWQQQHYSRTFPSGSLIVNPQRWNQDVQELRAATLGDARTVDVFGLYRQNTREKLNSAYDPTPYLSSTGYTTAETLAAYSDLTWHLTVRFSHDKSSTQY |
| (a) | ID1 is the number of template residues identical to query divided by number of aligned residues. |
| (b) | ID2 is the number of template residues identical to query divided by query sequence length. |
| (c) | Cov is equal the number of aligned template residues divided by query sequence length. |
| (d) | Norm. Zscore is the normalized Z-score of the threading alignments. A Normalized Z-score >1 means a good alignment and is highlighted in bold. |
| (e) | Download alignment lists the threading program used to identify the template, and provide the 3D structure of aligned regions of threading templates (threading[1-10].pdb.gz). |
| (f) | Template residues identical to query sequence are highlighted in color. |
|