| Rank | PDB
hit | ID1 | ID2 | Cov | Norm.
Zscore | Downloadalignment | | 20 40 60 80 100 120 140 160 180 200 220 240 260 280 300 320 340 360 380
| | | | | | | | | | | | | | | | | | | |
|---|
| SS
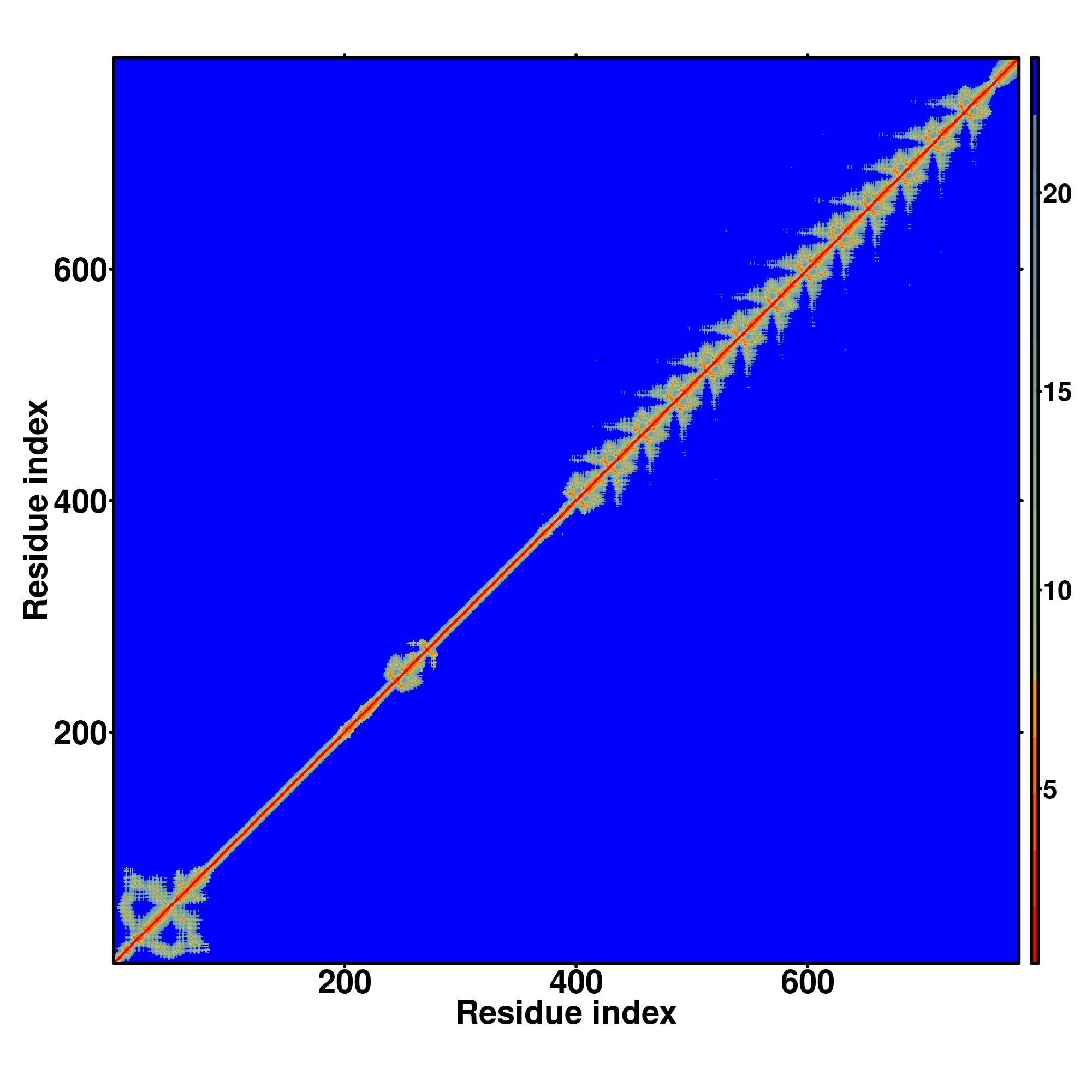
Seq | CCCCCCCCSSSSSSCSSCCHHHHHCCCHHHHHHHHHHHHHHHHCHHSSCCCCCCCCCCCHHHCCCCCCSSSSSCCCCCCCCCCCCCCCCCCCHHHHCSCCCSSSSSCCCCCCCCCCCCCCCCSSCCCCCCCCCCCCCCCCCSSCCCCCCCCCCCCCCCCCSSCCCCCCCCCCCCCCCSSSCCCCCCSCCCCCCCSCCCCCCCHHHCCCCCCCCCCCCCCCSCCCCCCCCSCCCCCCCCCSCCCCCCCSSCCCCCHHHSCCCSCCCCCSCCCCCCCSSCCCCCHCHSCCCSCCCCCSSCCCCCCSCCCCCCCCCSSCCCCCCCCSCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCSSSCCCCCCCCSCCCCCCCCCCC
MAEPPRLPLTFEDVAIYFSEQEWQDLEAWQKELYKHVMRSNYETLVSLDDGLPKPELISWIEHGGEPFRKWRESQKSGNIICSSVDMHFDPGFEEQLFWGSQQAMNSGKTKSHFQLDPESQCSFGSFVSFRPDQGITLGSPQRHDARAPPPLACGPSESTLKEGIPGPRNLDLPGLWDVPAWESTQHPWPVCGESCWENNHLVMHQRGHSKDRTRRAWEKFNKRAETQMPWSSPRVQRHFRCGVCGKSFRRKLCLLRHLAAHTGRGPFRNADGEMCFRHELTHPSHRLPQQGEKPAQCTPCGKRSLPVDSTQARRCQHSREGPASWREGRGASSSVHSGQKPGSRLPQEGNSHQEGDTEALQHGAEGPCSCSECGERSPM |
|---|
| 1 | 3ikmD | 0.06 | 0.05 | 2.37 | 0.75 | EigenThreader | | SSVNSLAEVHRLYPPLEKEPIRENFQDLMQYCAQDVWATHEVFQQQLPLFLERCPLKGTTELLPKRPQHLPGHPGWYRKLCPRLDDPAWTPGPSLLSLQMRVTPKLMALTWDGFPLH----YSERHGWGYLVPGRRDNLAKLESAGVVCPYHCLEQGKQQLMPQEARAAVPGQPLALTARGGPKDTQPSYHHGNGPYNDVDIPGCWFFKLPHKDGNSCNVGSPPDYDEEGLYGAILPQVVTAGTITRRAVEP------TWLTASNARPDR----VGSELKAMVQAPPGELWIAAVLGDAHFGCTAFGWMGEWLVRELNLPVDRTEGGWISLQ-----DLRKVQRETARKSQWKKWEVVAERAWKLESIATSDIPRTPVEE |
| 2 | 6bcuA | 0.05 | 0.04 | 1.74 | 0.83 | DEthreader | | ------------AGDTFTAEYVEFEVKALLFFFQQVCLTTQREPKEM-----GAARRWWLRRLSLRVTTLLNLAEMEPLPL------R-DDNGIVLLGERAAKCRAY-AKALHKEFPTPAILE--SL-ISINNKLQQPEAAGVLEYAMEQWYEKLHEW---EMLGRMLQCCEALGQ---------------A------VLGVDPSRLAYMKNMWKAKKEWQLNLQGINMEAVLHYKHQNQD-KKVTEDLSLLYGHWP-------------------PRPLVGRLIH--------LTVASKSTTTRNAANKIL---DLRQDERVLFLVTLLVLKVMLEAFVAIIRVRDKLTGRD-----LDV-QV--LIQATLCWCP---- |
| 3 | 5v3jE | 0.21 | 0.13 | 4.28 | 2.13 | FFAS-3D | | -----------------------------------------------------------------------------------------------------------------------------------PHKCKECGKAFHTPSQLSHHQKLHVGEKPYKCQECGKAFPSNAQLSLHHRVHTKCFECKECGKAFMRPSHLLRHQRIHTGEKPHECGKAFRYDTQLSLHLLTHAGARRFECKDCDKVYSCASQLALHQMSHTGEKPHKCKECGKGFISDSHLLRHQSVHTGETPYKCKECGKGFRRGSELARHQRAHSGDKPYKCKECGKSHQKVHTGDRPCKECGKAFIRRSELTHHERSHSGEKPYECKECGKTFG- |
| 4 | 1vt4I3 | 0.09 | 0.09 | 3.38 | 2.03 | MapAlign | | ------SILNTLQQLKFY-KPYICDNDPKYERLVNAILDFLPKGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGG--GGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGG---GGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGG |
| 5 | 5v3mC | 0.23 | 0.12 | 3.64 | 2.82 | CNFpred | | -----------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------CFECKECGKAFMRPSHLLRHQRIHTGEKPHKCGKAFRYDTQLSLHLLTHAGARRFECKDCDKVYSCASQLALHQMSHTGEKPHKCKECGKGFISDSHLLRHQSVHTGETPYKCKECGKGFRRGSELARHQRAHSGDKPYKCKECGKSFTCVHTGDRPHKCKECGKAFIRRSEHHERSHSGEKPYECKECGKTFGR |
| 6 | 3tspA | 0.06 | 0.06 | 2.69 | 1.24 | MapAlign | | ------PVACPECGPYLEWVSHGEHAEQEAALQAAIAQLKNIVAIKGIGGFHLACAVATLRARKHRPDAARQLLTTPAAPIVLVDKIAPGLNEVGVMLPANPLQHLLLVMTSGNLSGKPPAISNEQALEDLQGIADGFLIHNRDIVQRMDDSVVRESGEMLRRSRGYVPDALALPPGFKNVPPVLCLGADLKNTFCLVRGEQVVLSQHLGDLSDDGIQTQWREALRLMQNIYNFTPQYVVHDAHPGYSCLPTQTVHHAHALDGGDVIALTLWGGECLRVNYRECEHLGGL-PAVALPAAPWRNALASSCGRLFDAVAAHPVTMPRVDNQLDLATFAWAFHDALAQGFAALMREQATMRGITTLVFSGGVIHNRLLRAGDG |
| 7 | 5v3jE | 0.21 | 0.13 | 4.28 | 2.28 | MUSTER | | -----------------------------------------------------------------------------------------------------------------------------------PHKCKECGKAFHTPSQLSHHQKLHVGEKPYKCQECGKAFPSNAQLSLHHRVHTDEKCCKECGKAFMRPSHLLRHQRIHTGEKPHECGKAFRYDTQLSLHLLTHAGARRFECKDCDKVYSCASQLALHQMSHTGEKPHKCKECGKGFISDSHLLRHQSVHTGETPYKCKECGKGFRRGSELARHQRAHSGDKPYKCKECGKRHQKVHTGDRPHK-CKECGKAFIRRTHHERSHSGEKPYECKECGKTFGR |
| 8 | 6eudA | 0.06 | 0.06 | 2.55 | 1.13 | MapAlign | | ------WLPLQLLAHPGINGKIILLERRLAARNVAQRLAELVGYRMRLEVVTEGVLTRMIQRQADLALALLLDVRIGSDVLLCPLYGALSLNDQRKAIRVARFDPRTGLTRLITQRVSQASMTQRAGRAGRLEPGISEPEILQSDLSGLLMELLQWGCSDPAQMSWLDQPPVVNLLAAKRLLQMLGALEGERLSGMAPRLAAMLVQRSQQLLKRLNVRGGEADSSLIAPLLAGAFADRIARRQDGRYQLANGMGAMLDAALSIAPLLLQGSASPDARILLAIDELVQRCPQLVQQSDTVEWAWRRLQLHQAMLNGRGLLDWGMQQRLDSELPAHYTVPTGSRIAIRYHEDNPPALAVRMQEMFGEATNPTIAQGRVPLLS |
| 9 | 5v3jE | 0.23 | 0.13 | 4.20 | 1.76 | HHsearch | | ------------------------------------------------------------------------------------------------------------------------------------------------------PHKCKECGKA----FHTP--SQLSHHQK-LHVGEKPYKCQECGKAFPSNAQLSLHHRVHTDEKCFECGKAFMRPSHLLRHQRIHTGEKPHKCKECGKAFRYDTQLSLHLLTHAGARRFECKDCDKVYSCASQLALHQMSHTGEKPHKCKECGKGFISDSHLLRHQSVHTGETPYKCKECGKGFQRAHSGDKPYK---CKECGKSFTCTTELVHTGDRPHKCKECGKAFIR |
| 10 | 5v3jE | 0.17 | 0.11 | 3.74 | 1.11 | MapAlign | | ---------------PHKCKECGKAFHTPSQLSHHQKLHVGEKPYKCQECGKAFLSLHHRVHTDEKC-----FECKECGKAFMRPSHLLR--HQRIHTGEKPHKCKECGKAFRY----------------------------DTQLSLHLLTHAGARRFECKDCDKVYSCASQLALHQMSHTGEKPHKCKECGKGFISDSHLLRHQSVH-------------------------TGETPYKCKECGKGFRRGSELARHQRAHSGDKPYKCKECGKSFTCTTELFRHQKVHTGDRPHKCKECGKAFIRRSELTHHERSH---------------------------------------------SGEKPYECKECGKTFGR |
| (a) | ID1 is the number of template residues identical to query divided by number of aligned residues. |
| (b) | ID2 is the number of template residues identical to query divided by query sequence length. |
| (c) | Cov is equal the number of aligned template residues divided by query sequence length. |
| (d) | Norm. Zscore is the normalized Z-score of the threading alignments. A Normalized Z-score >1 means a good alignment and is highlighted in bold. |
| (e) | Download alignment lists the threading program used to identify the template, and provide the 3D structure of aligned regions of threading templates (threading[1-10].pdb.gz). |
| (f) | Template residues identical to query sequence are highlighted in color. |
|