| Rank | PDB
hit | ID1 | ID2 | Cov | Norm.
Zscore | Downloadalignment | | 20 40 60 80 100 120 140 160 180 200
| | | | | | | | | | |
|---|
| SS
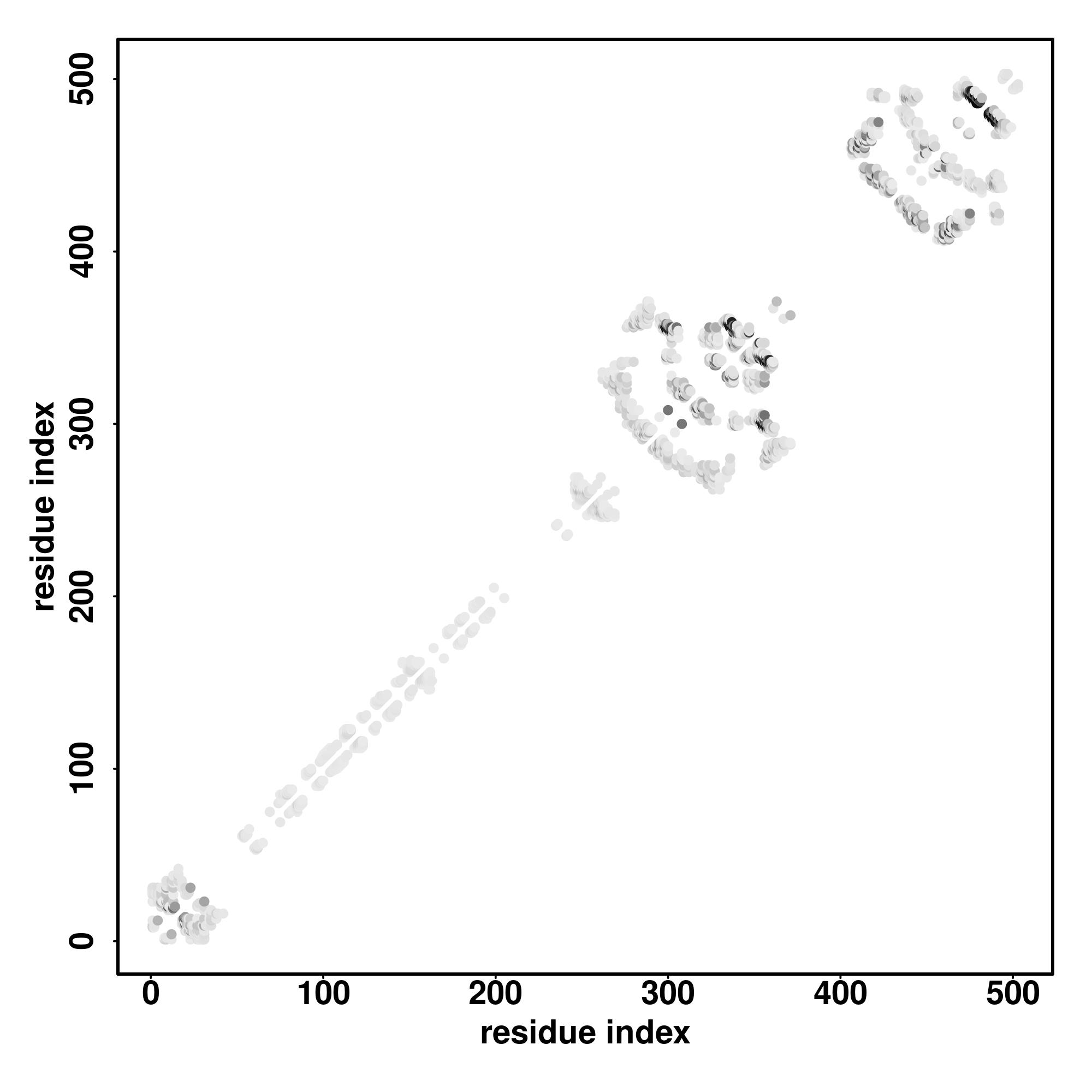
Seq | CCCCCHHHHHHHHHHHCCCCCCCCCHHHHHHHHHHHHHHHCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCC
MAGLSDLELRRELQALGFQPGPITDTTRDVYRNKLRRLRGEARLRDEERLREEARPRGEERLREEARLREDAPLRARPAAASPRAEPWLSQPASGSAYATPGAYGDIRPSAASWVGSRGLAYPARPAQLRRRASVRGSSEEDEDARTPDRATQGPGLAARRWWAASPAPARLPSSLLGPDPRPGLRATRAGPAGAARARPEVG |
|---|
| 1 | 3jc8Na | 0.07 | 0.07 | 2.82 | 1.05 | SPARKS-K | | RAVKKREMGRQVLVLFAYLWYDDRQSELEAHQAGVASTKARIAELEKIIGEVKNINTRKAEVEKKLAVLDALRKGRSGPVRMMDKKVWVKTSENNNAVSIDGSAVSHDEVAEFMRGLNGVVWTPKGMGRLVDRRRDSKTARVEMLT--------SDATIEEFPEAQVSPFFKNIDLQTAKQVGGAQVGVPILVEFKITMTSNY |
| 2 | 2bklB | 0.08 | 0.08 | 3.10 | 1.03 | MapAlign | | YPATRAEQVV--DTLHGVQVADP---YRWLEDEKAPEVQTWMTAQNAHAREALAKFPGREALAARFKELFTTFLQSDLSRDGKYLFVYILRGWSENDVYWKRPGEKDFRLLVKGVGAKYEVHAWKDRFYVLTDEGAPRQRVFEVDPLLSVSIVGGHLSLEYLKDATSEVRVATLKGKPVRTVDDAYYVFTSFTTPRQIYKTSV |
| 3 | 1jeiA | 0.27 | 0.07 | 2.11 | 2.46 | HHsearch | | YADLSDTELTTLLRRYNIPHGPVVGSTRRLYEKKIFEYETQRRRLSP-PSSS------------------------------------------------------------------------------------------------------------------------------------------------------- |
| 4 | 1ub2A1 | 0.10 | 0.10 | 3.66 | 0.54 | CEthreader | | WADLIAYAGTIAYESMGLKTFGFAFGREDIWHPEKDIYWGPEKEWFPPSTNPNSRYTGDRELENPLAAVTMGLIYVNPEGVDGNPDPLKTAHDVRVTFARMAMNDTVALTAGGHTVGKCHGNGNAALLGPEPEGADVEDQGLGWINKTQSGIGRNAVTSGLEGAWTPHPTQWDNGYFAVCSLNYDWELKKNPAGAWQWEPINP |
| 5 | 4r89A | 0.09 | 0.08 | 3.23 | 0.48 | EigenThreader | | ELPEASQALLVRMVMRKGTLAYAEIGDTRAAVQPLLALGAQLFGLLKKDELSQLFRKDALLERLFVLADLGIYRYESVEFSADSRGFRCRQRFDLGEPYANPWLEGRRVKLLFQFAQHCEKQRDFDLAQRLYR---QSSHPGARLRAIRSLERGERFAEAHALAREASCAPESDAERQGLARLLPRLQGKLGLP----RQARA |
| 6 | 1jeiA | 0.32 | 0.06 | 1.93 | 0.68 | FFAS-3D | | YADLSDTELTTLLRRYNIPHGPVVGSTRRLYEKKIFEYETQ------------------------------------------------------------------------------------------------------------------------------------------------------------------ |
| 7 | 7abiM | 0.07 | 0.07 | 2.99 | 1.00 | SPARKS-K | | GKSNYQLWHELCDLISQNPDKVQSLNVDAIIRGGLTRSLADYYIRRDVYEEAIRTVMTVRDFTQVFDSYAQFEESMIAAKMETASELGREEEDDVDLELRLARFEQLISRRPLLLNSVLLRQNPHHVHEWHKRVALHQGRPREIINTYTEAVQTVDVAFAKFYEDNGQLDDA-RVILEKATKVNFKQVASVWCQCGELELRHE |
| 8 | 2odcI | 0.32 | 0.07 | 2.08 | 0.65 | CNFpred | | YADLSDTELTTLLRRYNIPHGPVVGSTRRLYEKKIFEYETQRRR--------------------------------------------------------------------------------------------------------------------------------------------------------------- |
| 9 | 5uj9A3 | 0.05 | 0.03 | 1.60 | 0.67 | DEthreader | | SAVYVLAF-SLAHVASLVYSLWTHTVRLSVYGALGISQGITVFGYSMAVSIGGIFASRRLHLDLLHNVLRSPIFFEE--GLRYLVLKHI----------------GRTGKSTIPQDPLLSLAHLKG---------------------SA---------------D-QSTIRFDCTVL--T-IAHIYVIVL--EIQEW------ |
| 10 | 6yxaA | 0.06 | 0.06 | 2.58 | 0.87 | MapAlign | | ---IHPIQVAGILVDPSTIGFLHDVIRVILIKLADRLHNYRIVNLMKKKRAERELYVDEVVNEVKKRVEEVNIKADFSGRPKHIYSIYRKMVLQNKQFNEIYDLLAVRILVNSIKDCYAVLGIIHTCWKTVIGPKGDPLEVQIRFSDMVYVFTPKGDVIELPSGSVPIDFSYRIHSEIGNKTIGAKVNGHKLRTGDIVEILTS |
| (a) | ID1 is the number of template residues identical to query divided by number of aligned residues. |
| (b) | ID2 is the number of template residues identical to query divided by query sequence length. |
| (c) | Cov is equal the number of aligned template residues divided by query sequence length. |
| (d) | Norm. Zscore is the normalized Z-score of the threading alignments. A Normalized Z-score >1 means a good alignment and is highlighted in bold. |
| (e) | Download alignment lists the threading program used to identify the template, and provide the 3D structure of aligned regions of threading templates (threading[1-10].pdb.gz). |
| (f) | Template residues identical to query sequence are highlighted in color. |
|