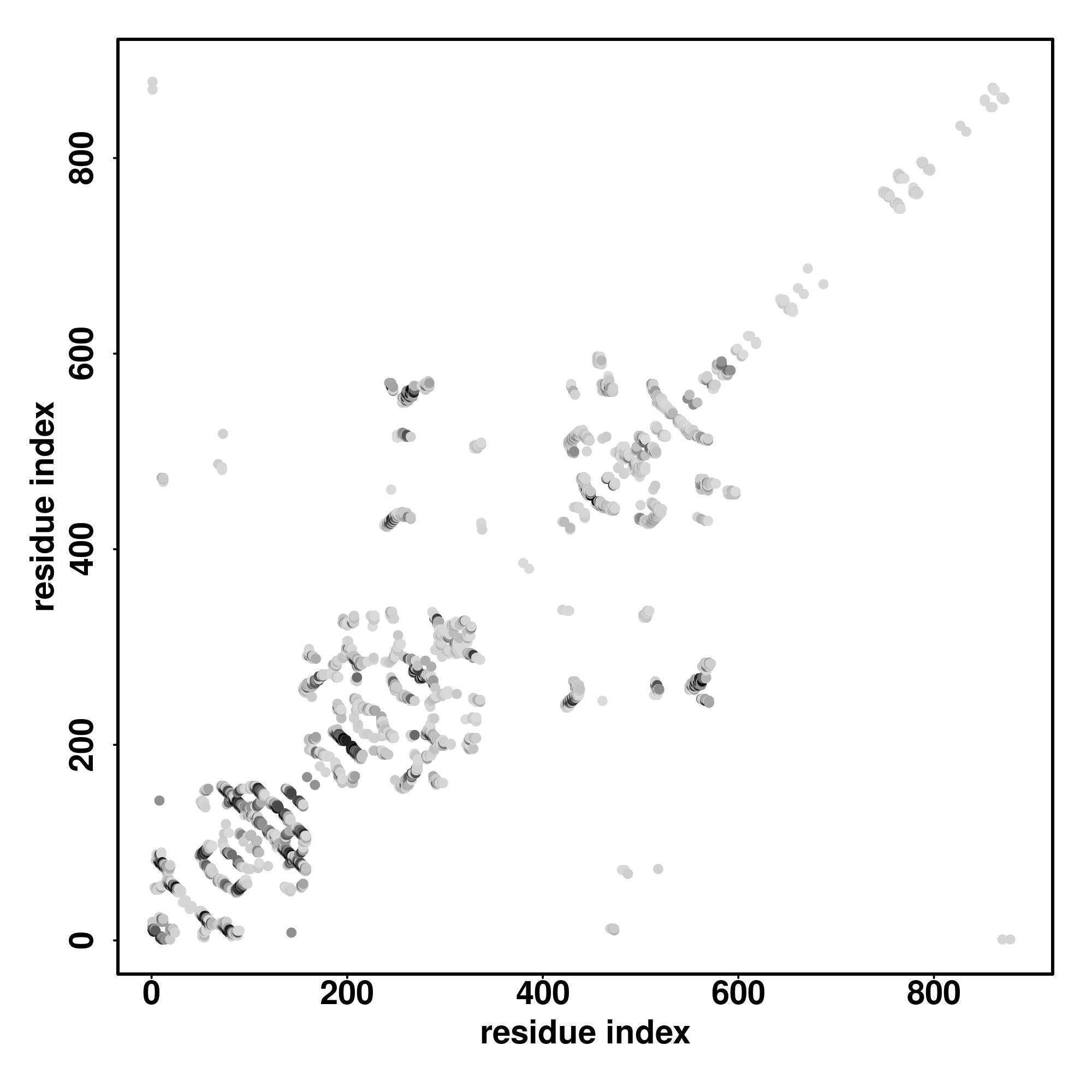
| Rank | PDB
hit | ID1 | ID2 | Cov | Norm.
Zscore | Downloadalignment | | 20 40 60 80 100 120 140 160
| | | | | | | | |
|---|
| SS
Seq | CCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCC
HRPAGSLAPSPAPTSSGPASSHKLGSCLLPDSFNIPGSSCSLLSSGDKPEAVMVIGKGLLGTGARMPCIKTRLQARPRLGRGSPPTRRGMKGSSGPTSPTPRTRESSELELGSCSATPGLPQARPPRPRSAPAFSPISCSLSDSPSWNCYSRGPLGQPEVCFVPKSPPLTVSPRV |
|---|
| 1 | 6l1eA | 0.10 | 0.10 | 3.59 | 0.69 | CEthreader | | -----GDAICIFRELQCLTPRGRYDIRIYPTFLHLHGKTLFLLPHKDQRQMFFVISLDPPITRYHFLILLFSKDEDISLTLNMNEEMKALVNRKITVPGNFQGHSGAQCITCSYKASSGLLIYVHKPPVHIRFDEISFVNFARGSFDFEIETKQGTQYTFSSIEREEYGKLFDFV |
| 2 | 2eidA | 0.12 | 0.11 | 4.07 | 0.60 | EigenThreader | | SYRNDAFGGSPGGIVTKHDMFCPGNDAKKPDMQVARGYQSSATMS---DGRVFTIGGSGSGGVFEKNGEVYSPLPNAKVNPMLTADKQGGPSTSGSGPDAMCGGSPDYQDSDATTNGLYFARTFQRRGSIVRVYHSISLLLPDGRVFNGGGGLCTNHFDAQIFTPNYLYNSNGNL |
| 3 | 3cnfB | 0.12 | 0.10 | 3.64 | 0.43 | FFAS-3D | | --PEGYVAVQYAHRLFSSSLANKRNR--VTYTHPPTGMAYP--SPTGRPHVHMTINESVIKSNWVVDILDIEVMTPSEGYTQHVDAESIMTAPKGKLFHLQFMDGLLRPEASGEDMRLIYPLQAIVNHNEVDRPREMDTGTLSRNGDLLYSPVANGQ------------------ |
| 4 | 7jjvA | 0.14 | 0.10 | 3.31 | 0.77 | SPARKS-K | | DGLDGADGSNGQAGASGLAGGPNCNGGKGGKGAPGVGTAGGAGGVGGAG------GTGNTNGGAGGSGGNSD-VAAGGAGAAGGAAGGAGTGGTGGNGGAGKPGG------APGAGGAGTPAGSAGSPGQTTVL----------------------------------------- |
| 5 | 5e9dE | 0.12 | 0.03 | 1.00 | 0.26 | CNFpred | | ---------------------------------------------------------------------------------------------------------------------------------------TLQCAQDMNEYMAWYRQDPGMGLRLIHYSVGVGITDQGDV |
| 6 | 6xs4A | 0.10 | 0.07 | 2.77 | 0.67 | DEthreader | | SIKAHKMAHIVKPADRPGAG----WLK-L-------SAGSPLYQNVPNLS----------------CGFGMPAFNNDEIVIPE----------AYDYATAVGYRCTGSFINFVMLALEGGRDAQILQVGTILGGVLL-Q-L-NPSTLDLLLFFKGWHIQYNIVSRETLLVRVAGY |
| 7 | 6w2uE1 | 0.07 | 0.06 | 2.31 | 0.87 | MapAlign | | ----------NHFNAYKLTRPYVAYCADCGMGHSCHSPAMIENIQADKIQFASQIGLTKTDTHDKIRYMGHFILA---------------------------KCPPGERISVSFVDSKNEHRTCRIAYHHEQRLITTYQLTETSEEIDMHMPPDSHTKWQFNKVHIPFPLI---- |
| 8 | 2nbiA | 0.24 | 0.24 | 7.46 | 0.76 | MUSTER | | -QPS-DLNPSSQPSECADVLEEPIDECFLPYSDASRPPSCLSFGRPDCDVLPTPQNINCPRCCA-TECRPDNPMFTPSPDGSPPICSPTMLPTNQPTPPEPSSAPSGEVIEECPLDTCFLPTSDPARPPDCTAGCPACCPFECSPDNPMFTPSPDGSPPNCSPTMLPPQPSTPTV |
| 9 | 2nrgA | 0.18 | 0.03 | 0.93 | 0.55 | HHsearch | | MLPIENQAPAPNITGALETGIELIKHLV--------------------------------------------------------------------------------------------------------------------------------------------------- |
| 10 | 2gcjC | 0.10 | 0.10 | 3.58 | 0.54 | CEthreader | | --EVAGDAIVSFQDVFFTTPRGRYDIDIYKNSIRLRGKTIVSLPKADDIHHLLVLAIPLRKGQTTYPFLVLQFQKDEETEVQLNLEDEDYEENYKDKLKKQYDAKTHIVLSHVLKGLTDRRVIVPGEYKSKYDQCAVSCSFKANEGYLYPL------DNAFFFLTKPTLYIPFSD |
| (a) | ID1 is the number of template residues identical to query divided by number of aligned residues. |
| (b) | ID2 is the number of template residues identical to query divided by query sequence length. |
| (c) | Cov is equal the number of aligned template residues divided by query sequence length. |
| (d) | Norm. Zscore is the normalized Z-score of the threading alignments. A Normalized Z-score >1 means a good alignment and is highlighted in bold. |
| (e) | Download alignment lists the threading program used to identify the template, and provide the 3D structure of aligned regions of threading templates (threading[1-10].pdb.gz). |
| (f) | Template residues identical to query sequence are highlighted in color. |
|