| Rank | PDB
hit | ID1 | ID2 | Cov | Norm.
Zscore | Downloadalignment | | 20 40 60 80 100 120 140 160 180 200 220 240
| | | | | | | | | | | | |
|---|
| SS
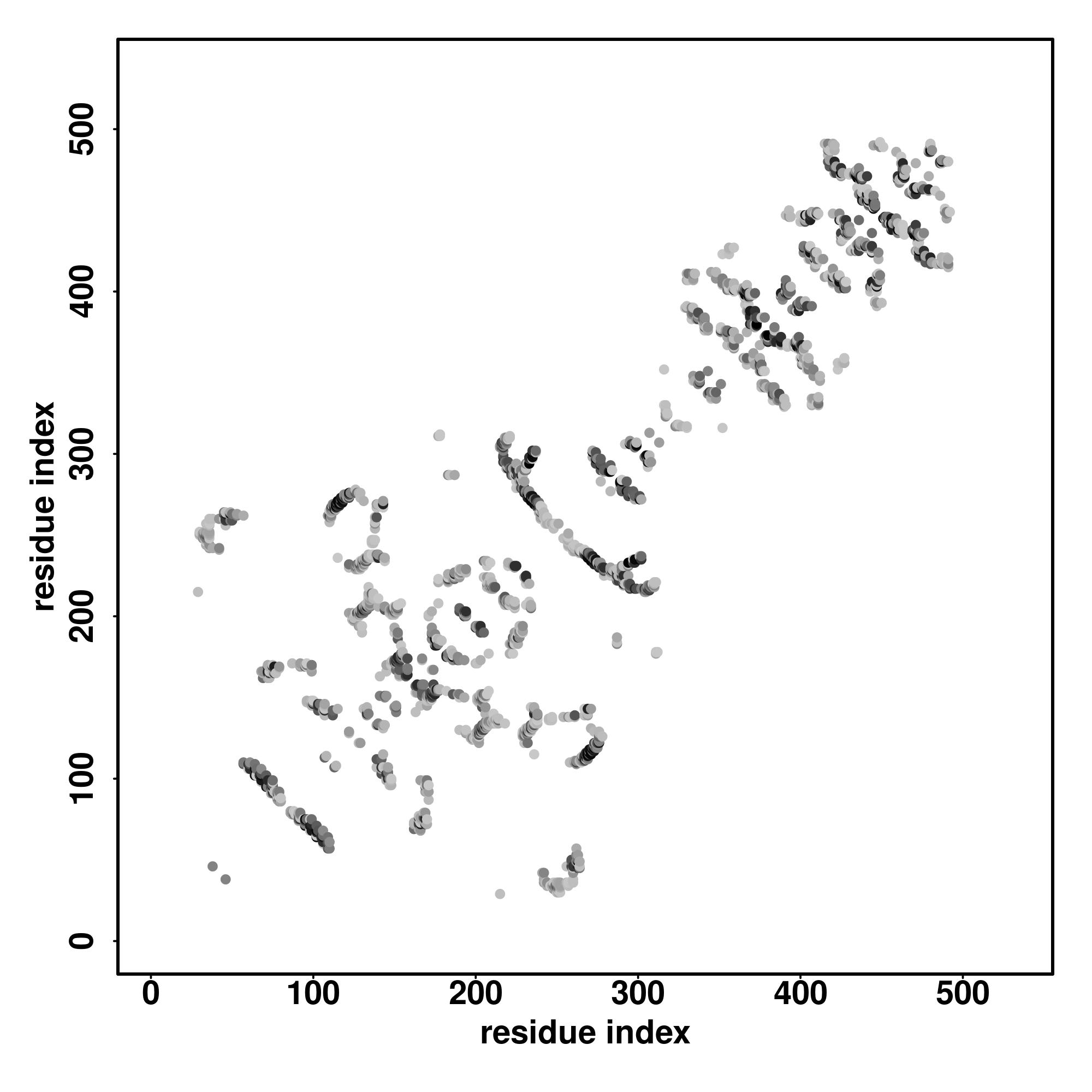
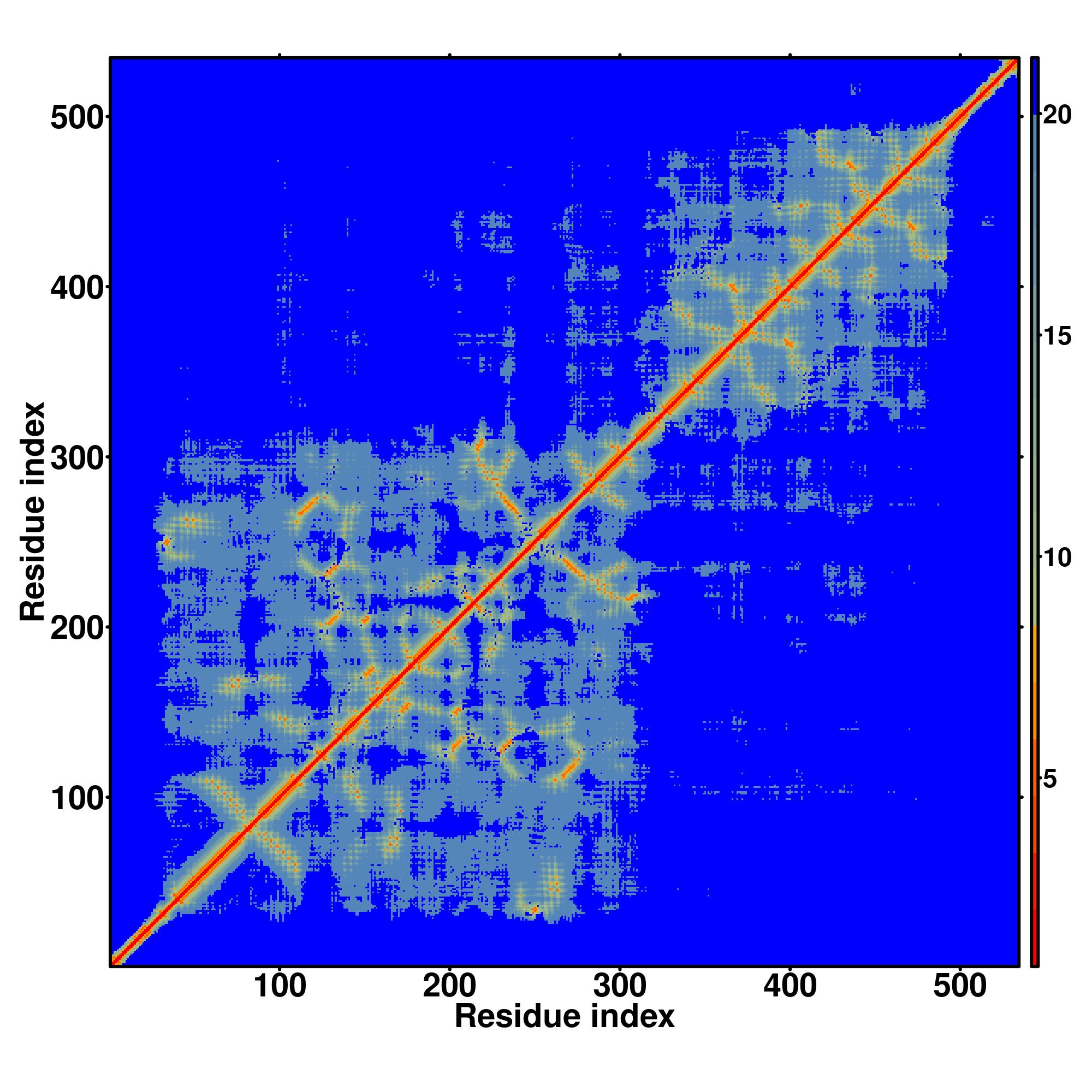
Seq | CCCCCCCCCCCCCCHHHHHHHHHCCCCCCHHHHHHHHHHHHHCCCCCCCHHHHHHHHHHHCCCHHHHHHHHHHHHHHCCCCCCCSSCHHHHHHHHCCCCCHHHHHHHHHHCCCCCCCSSHHHHHHHHHHHCCCCCHHHHHHHHHHHHCCCCCCCSCHHHHHHHHHHHHCCCHHHHHHHHHHHCCCCCCCSCHHHHHHHHHHCHHHHHHHHCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCC
MRLRGCGPRAAPASSAGASDARLLAPPGRFEDCQLALAEGQLRLPADTCLLEFARLVRGLGLKPEKLEKDLDRYSERARMKGGEKIGIAEFAASLEVPVSDLLEDMFSLFDESGSGEVDLRECVVALSVVCRPARTLDTIQLAFKMYGAQEDGSVGEGDLSCILKTALGVAELTVTDLFRAIDQEEKGKITFADFHRFAEMYPAFAEEYLYPDQTHFESCAETSPAPIPNGFCADFSPENSDAGRKPVRKKLD |
|---|
| 1 | 3dd4A | 0.12 | 0.09 | 3.38 | 1.23 | SPARKS-K | | NLEGLEMIAVLIVIVLFVKLLEQFGLIEDS----VEDELEMAT--VRHRPEALELLEAQSKFTKKELQILYRGFKN---ECPSGVVNEETFKEIYSGDSTTYAHFLFNAFDTDHNGAVSFEDFIKGLSILLRG-TVQEKLNWAFNLYDINKDGYITKEEMLDIMKAIYDMPRQHVETFFQKMDKNKDGVVTIDEFIESCQKDENIMRSMQLFENVI------------------------------------- |
| 2 | 3dd4A | 0.14 | 0.11 | 3.80 | 1.15 | MUSTER | | MNLEGLEMIVLIVIVLFVKLLE--QFGLIEDSVEDELEM----ATVRHRPEALELLEAQSKFTKKELQILYRGFKN---ECPSGVVNEETFKEIYSGDSTTYAHFLFNAFDTDHNGAVSFEDFIKGLSILLRG-TVQEKLNWAFNLYDINKDGYITKEEMLDIMKAIYDMPRQHVETFFQKMDKNKDGVVTIDEFIESCQKDENIMRSMQLFENVI------------------------------------- |
| 3 | 1g8iB | 0.19 | 0.12 | 3.97 | 1.68 | FFAS-3D | | ------------------------------------------KSNSKLKPEVVEELTRKTYFTEKEVQQWYKGFI---KDCPSGQLDAAGFQKIYFGDPTKFATFVFNVFDENKDGRIEFSEFIQALSVTS-RGTLDEKLRWAFKLYDLDNDGYITRNEMLDIVDAIYNTPEKRVDRIFAMMDKNADGKLTLQEFQEGSKADPSIVQALSLYD---------------------------------------- |
| 4 | 3dd4A | 0.14 | 0.11 | 3.63 | 1.09 | CNFpred | | ------------LFVKLLEQFGLI-----EDSVEDELEMATVRH----RPEALELLEAQSKFTKKELQILYRGFKNECP---SGVVNEETFKEIYS-DSTTYAHFLFNAFDTDHNGAVSFEDFIKGLSILLRG-TVQEKLNWAFNLYDINKDGYITKEEMLDIMKAIYD-PRQHVETFFQKMDKNKDGVVTIDEFIESCQKDENIMRSMQLFENVI------------------------------------- |
| 5 | 2r2iA | 0.16 | 0.10 | 3.41 | 0.83 | DEthreader | | ---------------------------------------------------------KAVELSATECHQWYKKFMTEC----PSGQLTYEFKQFFGLKNVEQMFETFDF--NKDGYIDF-MEYVAALSLVLKGKVD-QKLRWYFKLYDVDGNGCIDRGELLNIIKAIAIMTEEFTNMVFDKIDINGDGELSLEEFMEGVQKDEVLLDILTRLDL---------------THIVK--LIQNDG----------- |
| 6 | 5x9aA | 0.18 | 0.13 | 4.12 | 1.21 | SPARKS-K | | ------------------------------------------------NQKLAEELYKTSCFTKTEVESLIICYKNLL---EGLKMDRNLFRDILHMTEDLLMDRVFRAFDKDSDSYISLTEWVEGLSVFLRGTL-DEKMEYTFTVFDLNGDGYISREEMFQMLKTCLVPDKDLVEIALKKMDHDHDSRLSKKDFKDAVLIEPLLLEAFGKCLPDEKSSEIFEYHVLGVK----------------------- |
| 7 | 1uhnA | 0.18 | 0.12 | 3.98 | 0.71 | MapAlign | | ----------------------------------------------------PELLARDTVFSVSEIEALYELFKKISSVIDDGLINKEEFQLALFKTESLFADRVFDLFDTKHNGILGFEEFARALSVFHPNAPIDDKIHFSFQLYDLKQQGFIERQEVKQMVVATTVI-EDIIDKTFEEADTKHDGKIDKEEWRSLVLRHPSLLKNMTLQYLKDITTTFP------------------------------- |
| 8 | 1uhnA | 0.18 | 0.13 | 4.11 | 0.46 | CEthreader | | ---------------------------------------------------DPELLARDTVFSVSEIEALYELFKKISAVIDDGLINKEEFQLALFKTESLFADRVFDLFDTKHNGILGFEEFARALSVFHPNAPIDDKIHFSFQLYDLKQQGFIERQEVKQMVVATLAEIEDIIDKTFEEADTKHDGKIDKEEWRSLVLRHPSLLKNMTLQYLKDITTTFPSFVFH-------------------------- |
| 9 | 5x9aA | 0.18 | 0.13 | 4.13 | 1.07 | MUSTER | | ---------------------------------------------NQKLAEELYKTSCQKHFTKTEVESLIICYKNLL---EGLKMDRNLFRDILHMTEDLLMDRVFRAFDKDSDSYISLTEWVEGLSVFLRG-TLDEKMEYTFTVFDLNGDGYISREEMFQMLKTCLVGIKDLVEIALKKMDHDHDSRLSKKDFKDAVLIEPLLLEAFGKC-------LPDEKSSEIFEYHVLGVK---------------- |
| 10 | 3evuA | 0.14 | 0.11 | 3.75 | 0.91 | HHsearch | | SGEGEGDATYGKLMKFFKSAMPEGYIQERTIFF-----KDDGNYKTRAEVK-FELVNEYNTLTEEQIAEFKEAFSLF-DKDGDGTITTKELGTVMRSPTEAELQDMINEVDADGNGTIDFPEFLTMMARKMKDTDSEEEIREAFRVFDKDGNGYISAAELRHVMTNLEKLTDEEVDEMIREADIDGDGQVNYEEFVQMMTA---------------------------------------------------- |
| (a) | ID1 is the number of template residues identical to query divided by number of aligned residues. |
| (b) | ID2 is the number of template residues identical to query divided by query sequence length. |
| (c) | Cov is equal the number of aligned template residues divided by query sequence length. |
| (d) | Norm. Zscore is the normalized Z-score of the threading alignments. A Normalized Z-score >1 means a good alignment and is highlighted in bold. |
| (e) | Download alignment lists the threading program used to identify the template, and provide the 3D structure of aligned regions of threading templates (threading[1-10].pdb.gz). |
| (f) | Template residues identical to query sequence are highlighted in color. |
|