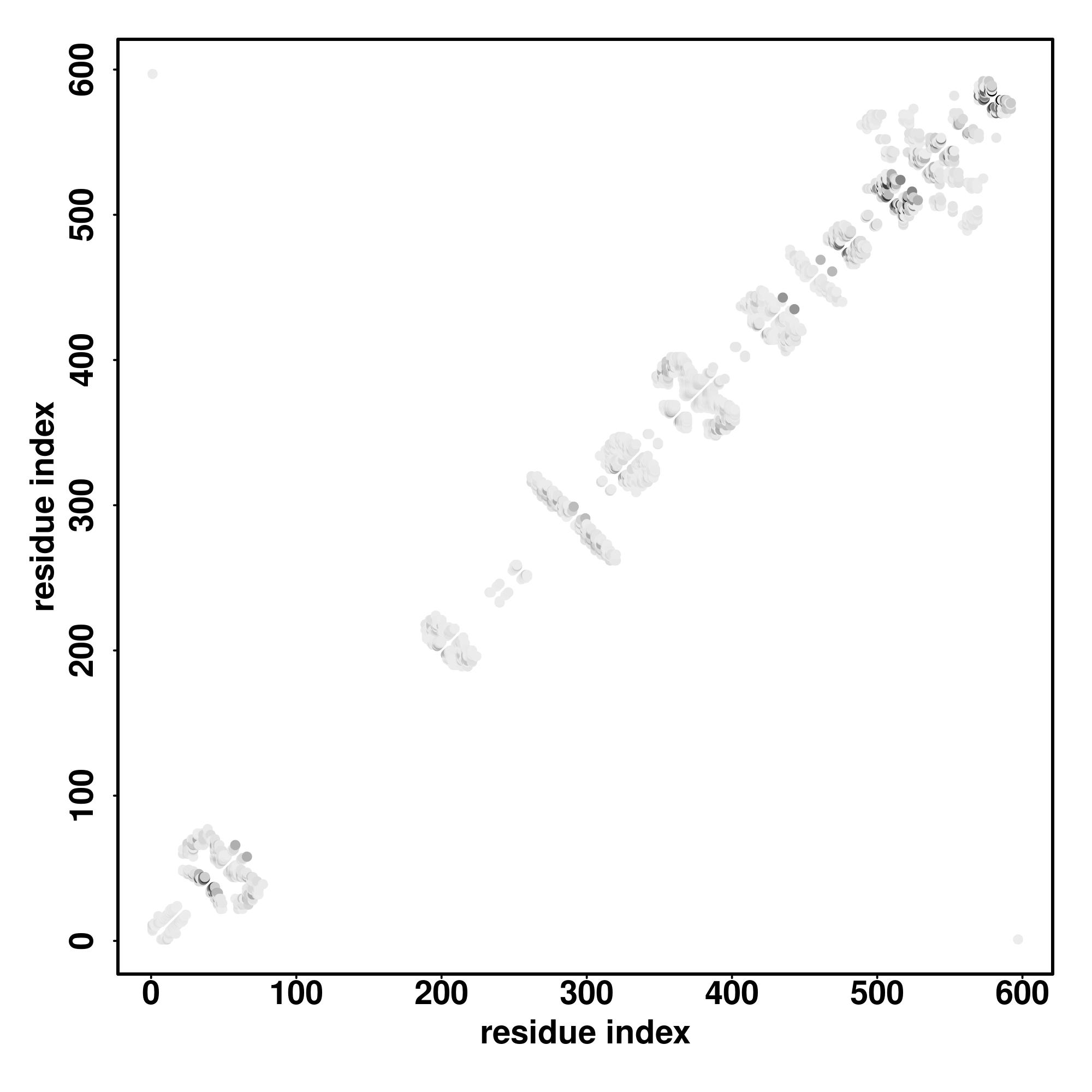
| Rank | PDB
hit | ID1 | ID2 | Cov | Norm.
Zscore | Downloadalignment | | 20 40 60 80 100 120 140 160 180 200 220 240
| | | | | | | | | | | | |
|---|
| SS
Seq | CCCCCCCCCCHHHHHHHHHHHHHHHHHHHHHHHHHHHHCCCCCCCCCCCCCCCHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHCCCCHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHCC
MFPVAPKPQDSSQPSDRLMTEKQQEEAEWESINVLLMMHGLKPLSLVKRTDLKDLIIFDKQSSQRMRQNLKLLVEETSCQQNMIQELIETNQQLRNELQLEQSRAANQEQRANDLEQIMESVKSKIGELEDESLSRACHQQNKIKDLQKEQKTLQVKCQHYKKKRTEQEETIASLQMEVCRLKKEEEDRIVTQNRVFAYLCKRVPHTVLDRQLLCLIDYYESKIRKIHTQRQYKEDESQSEEENDYR |
|---|
| 1 | 3na7A | 0.12 | 0.10 | 3.55 | 0.49 | CEthreader | | -------------------------------------------LGSNTHLKQLIEISHLDKEIDSLEPLIREKRKDLDKALNDKEAKNKAILNLEEEKLALKLQVSKNEQTLQDTNAKIASIQKKMSEELRSLNIEEDIAKERSNQANREIENLQNEIKRKSEKQEDLKKEMLELEKLALELESLVENEVKNIKETQQIIFKK--KEDLVEKTEPKIYSFYERIRRWAKNTSIVTIKKQACGGCFIR |
| 2 | 4l3iA | 0.09 | 0.09 | 3.38 | 0.85 | EigenThreader | | EEGETTILQLEKDLRTQVELRRKQELKLLQEQDQELCEILCPHYDIDSASVPS------LEELNQFRQHVTTLRETKASRREEFVSIKRQIILCFERDVVCENIATLQKLLRQLEQKSQNEAVCEGLRTQIRELWDRLQVIEAIRVELVQYWDQCFYSQEQRQLLQLHDAEIVRLKNYYEVHQKWEETWRLFLEFERKASFTNRGGNLLKEEKQRAKLQKLPKLEEELKARIELWEQEHYVAEQWEH |
| 3 | 5xg2A | 0.11 | 0.09 | 3.35 | 1.34 | FFAS-3D | | -------------------KRKEKLIEEIRAREEERNA--------------LVVRLGEIDRTFAVAREEDTVVKELEGEARIKREKERLKAEIEARLPGLRERAENLRRLVEEKRAEISELERRLSSITFELRIKLSDLEKELELARKDLEKVLAEERAVREEIEVAKRRINELDTLIERERGELAKLRGRIERLERKRDKKALENPEARELTEKIRAVEKEIAALREELSRVEGKLEG------- |
| 4 | 5xg2A | 0.14 | 0.13 | 4.30 | 0.97 | SPARKS-K | | -----------------TKGAIVRWGKRKEKLIEEIRAREEERNALVRLGEIDRTFAVAREEFDTVVKELEEARKSLYEGEARIKRAEEEKERLKAEILTGEARLPGLRERAENLRRLVEEKRAEISELERRLSSILSDLEKELELARKDLEKVLAEERAVREEIEVAKRRINELDTLIERERGELAKLRGRIERLERKRDKLALENPEARELTEKIRAVEKEIAALREELSRVEGKLEGL------ |
| 5 | 5j1iA | 0.11 | 0.08 | 2.95 | 0.72 | CNFpred | | ---------------SLVIRGTQGAEEVLRAHEEQLKEA--------------QAVPATLPELEATKASLKKLRAQAEAQQPTFDALRDELRGAQEVGERLQQRHGERDVEVERWRERVAQLLERWQAVLAQTDVRQ-----------RELEQLGRQLRYYRESADPLGAWLQDARRRQEQI-QAVREQLRQEQALLEEIERHG-CQRFAKQYINAIKDYELQLVTYKAQ----------------- |
| 6 | 2dfsA | 0.04 | 0.03 | 1.54 | 1.00 | DEthreader | | -----INAYSGQ-GDMD------TYQEFFSRYRVLMKQKDVQTCKN--------FGKTKIFF--GQVAYLEKIRADKLRAACIRIQKTIRGWLMRKKYMRMRRAAITIQRYVRGHQARCYATFLRRTRAAIIIQKFQRMYVVRKRYQCMRDATIALQALLRGYLVRNKYQMMLREHKSIIIQKHVRGWLARVHYHRTLKAIVYQC------------MNNLEITY-S-------------------- |
| 7 | 6rlbA | 0.09 | 0.09 | 3.28 | 0.82 | MapAlign | | ---LVKRPTISKELMLERETLLARLVDSIKDFRLDFEKVDDTIKIAEALLSDLPGFRCFHQSAKDLLDQLKLYEQEQFDDWSRDIQSGLRLVILLREVRQLSKIQQVANIAQKFCKQAIILKQVAHFYNSIDQQMMLQSALAFEQIIKNSLEGYIQKLQNAAERLATENRKLRKWHTTFCEKVVVLKDGLQELRTGLATVEAQGFQASDMHAWKQHWNHQLYKALEHQYQMGLEAPPFEEIRAKYYR |
| 8 | 5nnvA | 0.12 | 0.11 | 3.70 | 0.99 | MUSTER | | ----------AKEEELAESSAISAKEAKIEDTRDKIQAL--------------------DESVDELQQVLLVTSEELEKLEGRKEVLKERKKNAVQNQEQLEEAIVQFQQKETVLKEELSKQEAVFETLQAEVKQLRAQVKEKQQ-LSNELTELKIAAAKKEQACKGEEDNLARLKKELTETELALKEAKEDLSFLTS----ESSSTSGEEKLEEAAKHKLNDKTKTIELIALRRDQRIKLQHGLDT |
| 9 | 6mi3A | 0.17 | 0.08 | 2.65 | 0.68 | HHsearch | | -----------------------------------------------------------SWSVKELEDKNEELLNEVARLKKLLQRCLAANQELRDAIRQS-NQI--LRERAEELLHFQASQREEKEFLMSKF----QEARKLVERLGLEKLELEDKNEELLSEIAHLKNEVARLKKLVGE------------------------------------------------------------------ |
| 10 | 2tmaA | 0.10 | 0.10 | 3.73 | 0.48 | CEthreader | | LDRAEQAEADKKAAEDRSKQLEDELVSLQKKLKGTEDELDKYSEALKDAQEKLELAEKKATDAEADVASLNRRIQLVEEELDRAQERLATALQKLEEAEKAADESERGMKVIESRAQKDEEKMEIQEIQLKEAKHIAEDADRKYEEVARKLVIIESDLERAEERAELSEGKCAELEEEIKTVTNNLKSLEAQAEKYSQKEDKYEEEIKVLSDKLKEAETRAEFAERSVTKLEKSIDDLEDELYAQKL |
| (a) | ID1 is the number of template residues identical to query divided by number of aligned residues. |
| (b) | ID2 is the number of template residues identical to query divided by query sequence length. |
| (c) | Cov is equal the number of aligned template residues divided by query sequence length. |
| (d) | Norm. Zscore is the normalized Z-score of the threading alignments. A Normalized Z-score >1 means a good alignment and is highlighted in bold. |
| (e) | Download alignment lists the threading program used to identify the template, and provide the 3D structure of aligned regions of threading templates (threading[1-10].pdb.gz). |
| (f) | Template residues identical to query sequence are highlighted in color. |
|