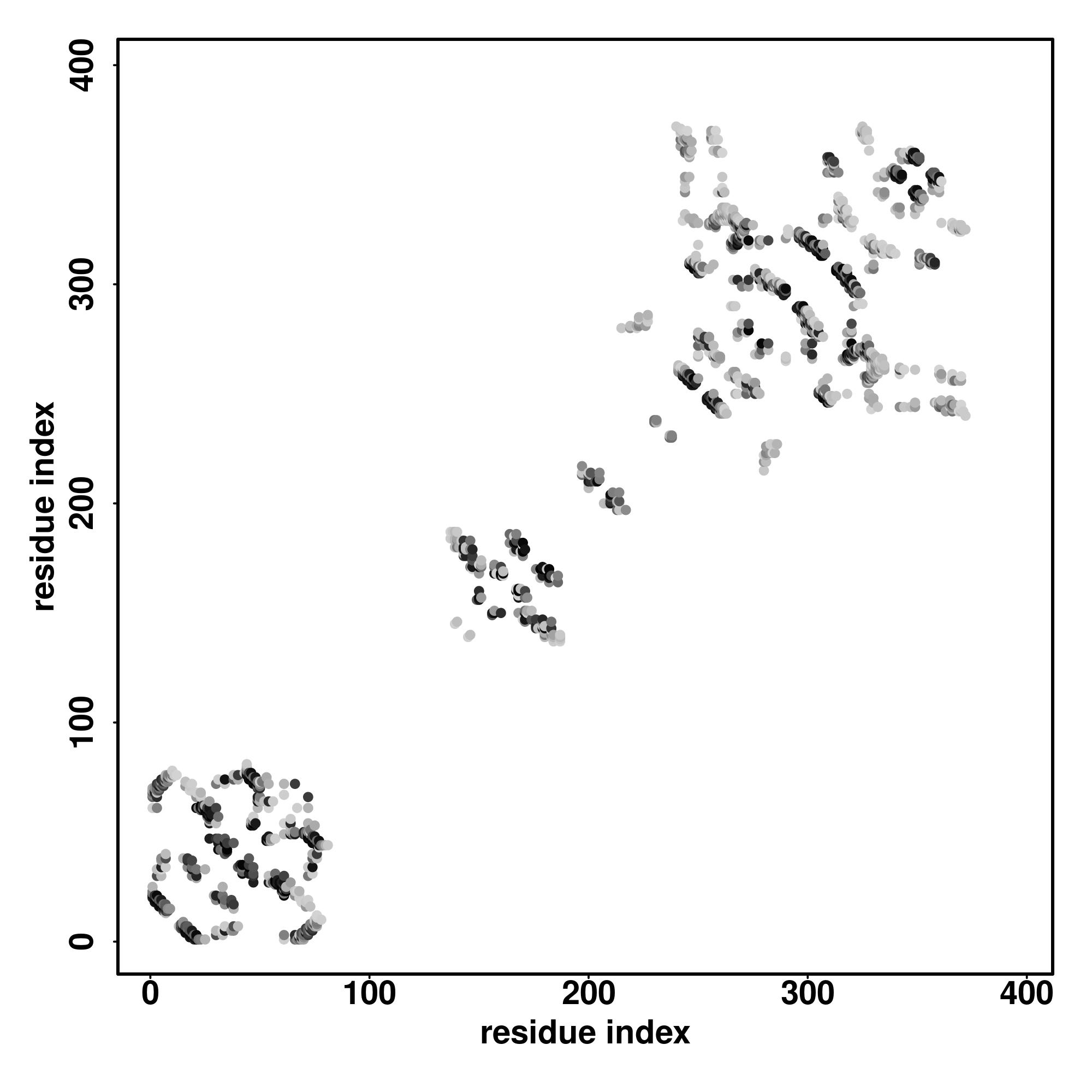
| Rank | PDB
hit | ID1 | ID2 | Cov | Norm.
Zscore | Downloadalignment | | 20 40 60 80 100 120 140 160 180 200 220 240 260 280
| | | | | | | | | | | | | | |
|---|
| SS
Seq | CCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCHHHHHHHHHCCHHHHHHHHHHCCHHHHHHHCCCHHHHHHHHHHHHHHHHHHHHHHHHHHCCCCCCHHHHHHHHHHHHHHHHHHHHHHHHHHCCCCCCCCCSSSSSSSSCCSSSSSSSCCCCCCCSCCHHHHHHHCCCCCCCCCCCCSSCCCCCCCSSSSSSSSSSSSCCSSSSSSSSSSCCCCCCSSSSHHHHHHHCCSSSCCCCSSSSCCCCSSSSSCCCCCCCCCCCCCCCCCCCCCHHHHHHHHHHCCCCC
TSSSRPQHPGQQQQRTPAAQRSQGLASGEKVAGLQGLGSPALIRSMLLSNPHDLSLLKERNPPLAEALLSGSLETFSQVLMEQQREKALREQERLRLYTADPLDREAQAKIEEEIRQQNIEENMNIAIEEAPESFGQVTMLYINCKVNGHPLKAFVDSGAQMTIMSQACAERCNIMRLVDRRWAGVAKGVGTQRIIGRVHLAQIQIEGDFLQCSFSILEDQPMDMLLGLDMLRRHQCSIDLKKNVLVIGTTGTQTYFLPEGELPLCSRMVSGQDESSDKEITHSVMDSGRKEH |
|---|
| 1 | 3s8iA | 1.00 | 0.42 | 11.85 | 1.14 | SPARKS-K | | -----------------------------------------------------------------------------------------------------------------------------------------VTMLYINCKVNGHPLKAFVDSGAQMTIMSQACAERCNIMRLVDRRWAGVAKG---QRIIGRVHLAQIQIEGDFLQCSFSILEDQPMDMLLGLDMLRRHQCSIDLKKNVLVIGTTGTQTYFLPEGELP----------------------------- |
| 2 | 5kesA | 0.26 | 0.09 | 2.84 | 1.44 | FFAS-3D | | ---------------------------PLGSATLSDEAFIEQFRQELLNNQMLRSQLILQIPGLNDLVN--DPLLFRERLGPLILQRRYIPQDEYTRLMANPDDPDNKKRIAELLDQQAIDEQLRNAIEYTPE---------------------------------------------------------------------------------------------------------------------------------------------------------------- |
| 3 | 3s8iA | 1.00 | 0.42 | 11.85 | 3.25 | HHsearch | | -----------------------------------------------------------------------------------------------------------------------------------------VTMLYINCKVNGHPLKAFVDSGAQMTIMSQACAERCNIMRLVDRRWAGVAKG---QRIIGRVHLAQIQIEGDFLQCSFSILEDQPMDMLLGLDMLRRHQCSIDLKKNVLVIGTTGTQTYFLPEGELP----------------------------- |
| 4 | 5kesA | 0.27 | 0.10 | 2.94 | 2.95 | HHsearch | | -----------------------GPL---GSATLSDEAFIEQFRQELLNNQMLRSQLILQIPGLNDLVN--DPLLFRERLGPLILQRRYIPQDEYTRLMANPDDPDNKKRIAELLDQQAIDEQLRNAIEYTPE---------------------------------------------------------------------------------------------------------------------------------------------------------------- |
| 5 | 3s8iA | 0.99 | 0.42 | 11.76 | 1.69 | FFAS-3D | | -----------------------------------------------------------------------------------------------------------------------------------------VTMLYINCKVNGHPLKAFVDSGAQMTIMSQACAERCNIMRLVDRRWAGVAKGQ---RIIGRVHLAQIQIEGDFLQCSFSILEDQPMDMLLGLDMLRRHQCSIDLKKNVLVIGTTGTQTYFLPEGELP----------------------------- |
| 6 | 4rghA | 0.93 | 0.40 | 11.12 | 1.01 | CNFpred | | -------------------------------------------------------------------------------------------------------------------------------------------MLYINCKVNGHPVKAFVDSGAQMTIMSQACAERCNIMRLVDRRWAGIAKGVGTQKIIGRVHLAQVQIEGDFLPCSFSILEEQPMDMLLGLDMLKRHQCSIDLKKNVLVIGTTGSQTTFLPEGELP----------------------------- |
| 7 | 5zcsE | 0.06 | 0.04 | 1.89 | 0.67 | DEthreader | | RIALTILLAEGGSLIGVDPGPMDNYL----PHSHS-HLHEMKPKPYSLKATHKRKDIFINYKDEQLHRFVYADLDKAKQLTVVGCQFTEFLLESEILDTLIQMLLRFSIPKGFSYLEATT--KP-DG------------------NQRLQR-----------------PHVYL-PI---------------------HLYGQHGCHLLEV--------LDKWEEIKLSLLGNIG-SSNWGL--NLLQ-------EENVIDILKLLSRGTCYVKWVRRKHLWPVVPD------- |
| 8 | 5yq8A | 0.48 | 0.21 | 6.16 | 1.14 | SPARKS-K | | ------------------------------------------------------------------------------------------------------------------------------------AKVTML---YVPCTINQVLVKAFVDSGAQNSIMNKRTAERCGLMRLVDVRMRGVAVGVGRQEICGRIHMTPVNLAGMYIPFAFYVIEDQAMDLIIGLDQLKRHQMMIDLKHNCLTID--NINVPFLPENDLPALA-------------------------- |
| 9 | 3s8iA | 0.98 | 0.41 | 11.57 | 0.92 | MapAlign | | ------------------------------------------------------------------------------------------------------------------------------------------TMLYINCKVNGHPLKAFVDSGAQMTIMSQACAERCNIMRLVDRRWAGVA---KGQRIIGRVHLAQIQIEGDFLQCSFSILEDQPMDMLLGLDMLRRHQCSIDLKKNVLVIGTTGTQTYFLPEGELP----------------------------- |
| 10 | 3s8iA | 1.00 | 0.42 | 11.85 | 0.90 | CEthreader | | -----------------------------------------------------------------------------------------------------------------------------------------VTMLYINCKVNGHPLKAFVDSGAQMTIMSQACAERCNIMRLVDRRWAGVAKG---QRIIGRVHLAQIQIEGDFLQCSFSILEDQPMDMLLGLDMLRRHQCSIDLKKNVLVIGTTGTQTYFLPEGELP----------------------------- |
| (a) | ID1 is the number of template residues identical to query divided by number of aligned residues. |
| (b) | ID2 is the number of template residues identical to query divided by query sequence length. |
| (c) | Cov is equal the number of aligned template residues divided by query sequence length. |
| (d) | Norm. Zscore is the normalized Z-score of the threading alignments. A Normalized Z-score >1 means a good alignment and is highlighted in bold. |
| (e) | Download alignment lists the threading program used to identify the template, and provide the 3D structure of aligned regions of threading templates (threading[1-10].pdb.gz). |
| (f) | Template residues identical to query sequence are highlighted in color. |
|