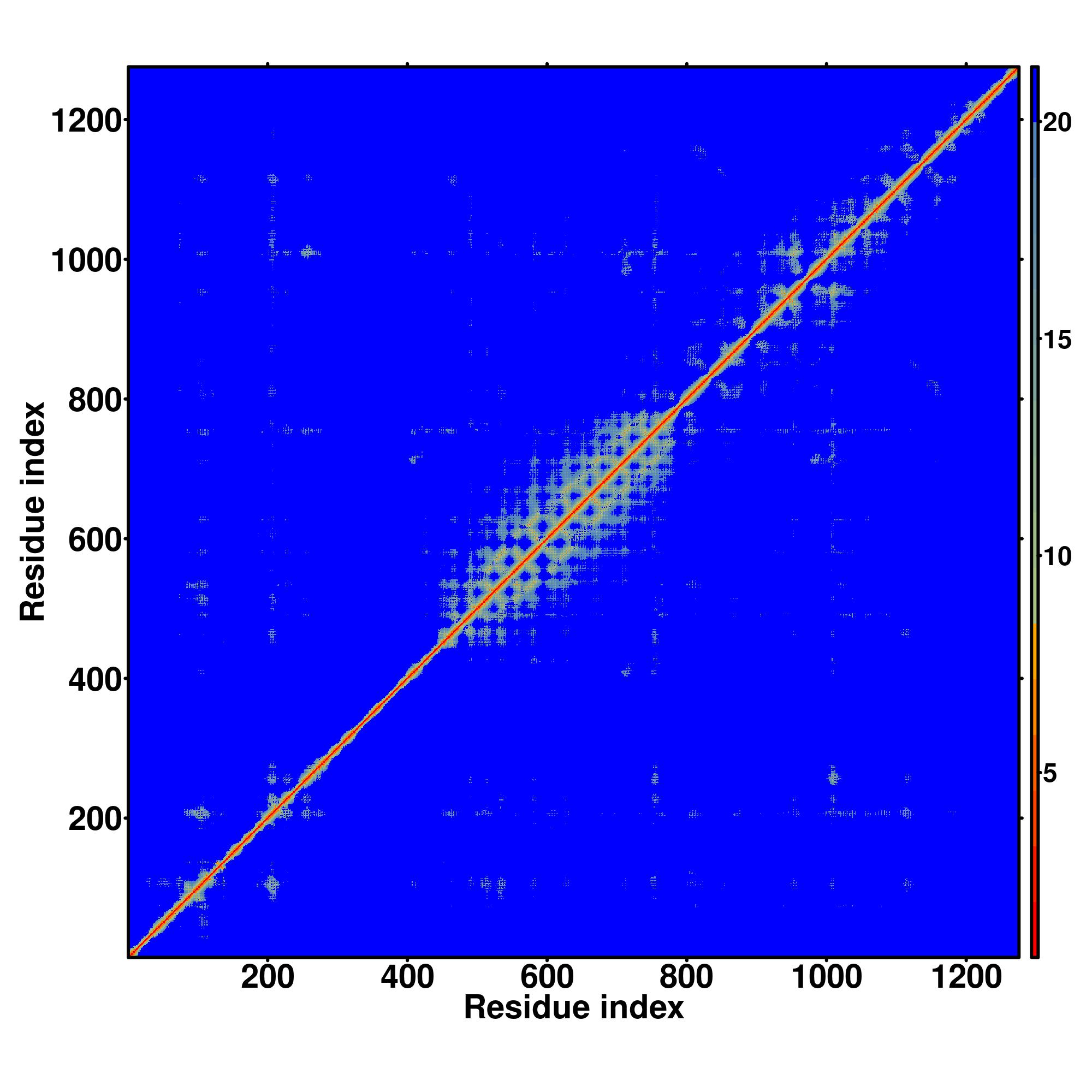
| Rank | PDB
hit | ID1 | ID2 | Cov | Norm.
Zscore | Downloadalignment | | 20 40 60 80 100 120 140 160 180
| | | | | | | | | |
|---|
| SS
Seq | CCCHHHHHHHHHHHHCCCHHHHHHCHHHHHHHHHHHHHHHHHHHHHHCCCCCHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHCCCCCCCCCCCCCCCCCCHHHHHHHHHHCHHHHHHHCHHHHHHHHHHHHHHHHCHHHHHHCHHHHHHHHHHHHHHHHHCCCCCHHHHHHHHHHHHHHHHHCCCCCCC
DQDWNDFLQQVCSQIDSTEKSMGASRAKLNLLCYLCVVAGHQEVARKTTPGKEQQSGNEYLSKCLDLLICHIVQELPRILGDILNSLANVSGRKHPSTVQVKQLKLCLPLMPVVLHLVTSQVFRPQVVTEEFLFSYGTILSHIKSPILLKDYRSTVVDYILPPLVSLVQSQNVEWRLFSLRLLSETTSLLVNQEFGDG |
|---|
| 1 | 1qgkA1 | 0.08 | 0.08 | 3.04 | 0.43 | CEthreader | | ANARREVKNYVLHTLGTETYRPSSASQCVAGIACAEIPVLIPQLVANVTNPNSTEHMKESTLEAIGYICQDIDPEQDKSNEILTAIIQGMRKEEPSNNVKLAATNALLNSLEFTKANFDKESERHFIMDTRVRVAALQNLVKIMS-LYYQYMETYMGPALFAITIEAMKSDIDEVALQGIEFWSNVCDEEMDLAIEAS |
| 2 | 7kflA | 0.08 | 0.08 | 3.16 | 0.63 | EigenThreader | | HLNEKQQENQDLLVKCISQDRIIQTIATAIEEVLAYWLSNSATLLLLLQRRQGLTKLDDQLTAFLEKIYGMIRDNLKKEISPLLGLCIQAPRTSRNAVAQQALIAHWQSIRSYLNLMKANNFLVRKVFTQIFSFINVQLFNSLLCSFSNGEYVKAGL----AELEQWCIEATDEYAGSAWDELRHIRQAVGFLVIHQK |
| 3 | 1b3uA3 | 0.16 | 0.13 | 4.38 | 0.93 | FFAS-3D | | RQLSQSLLPAIVELAEDAKWRVRLAIIYMPLLAGEKLNSLCMAWL---------VDHVYAIREAATSNLKKLVEKFEWAHATIIPKVLAMSGDPNYLHEVCGQDITTKHMLPTVLRMAGDPVAN---VRFNVAKSLQKIGPILD--------NSTLQSEVKPILEKLTQDQDVDVKYFAQEALTVL------------ |
| 4 | 6w2wA | 0.13 | 0.12 | 4.22 | 0.74 | SPARKS-K | | SEEVNERVKQLAEKAKEAEEVIEIVKELAELAKQAEIVYQLAEVAEHSTD-------PELIKEILQEALRLAEEQAARLALKAARLLEEARQSKDPE---NEAAKECLKAVRAALEAALLALLLLAKAAQDAVQLATAALRAVEACQLAKQYPKKCIKAASEAAEEASKAPDSQKARDEIKEASQKAEEVKERCERA- |
| 5 | 4gmxC | 0.06 | 0.06 | 2.42 | 0.82 | CNFpred | | YPQLGHIYYNMLQLYRAVSSMISAPKVRGLRTIKKEILKLVETYISKARNLDDV--VKVLVEPLLNAVLEDYDAEVLNCMTTVVEKVGHMIPQ------------GVILILQSVFECTL-DMINKDFTYPEHRVEFYKLLKVINSFAAFLELPPAAFKLFVDAICWAFKHNNRDVEVNGLQIALDLVKNIERMGNVPF |
| 6 | 3j7jC | 0.08 | 0.07 | 2.79 | 1.00 | DEthreader | | --RA-QI-ELLQLLVIAAENNGEGVIVKIKFICINELMDILFANPNIGEN-IL-EESEN--AD-PLRVRGCILTLVERMDEEFTKIMQNT-DP--HSQEYVEHLKDEAQVCAIIERVQRYEEKGTTEEVCRIYLLRILHTYY-KFDYKAHDSAVLMERLCKYAILMMSHLQDNIQ--LYNRTMQLG-CA---P----- |
| 7 | 3s4wA3 | 0.09 | 0.09 | 3.29 | 0.58 | MapAlign | | -PQFVQMLSWTSKICKEYSQEDASFCKSLMNLFFRDLSQDIHGQLGDIDQDVEIEKFAVVNLTAAPTVCLLVLSQAEKVLEEVDWLIAKIKGSAPT-LLIEKAIVMQLTLVTFFHELVQTALPGSCTL-LKGLSKIYSTLTAF--VKYYLQVCKLSGSHLTPVCYSFISQNKSSPIPNLVFAIEQYEKFLIQLSKKSK |
| 8 | 4uosA | 0.10 | 0.09 | 3.39 | 0.66 | MUSTER | | -GDNEEVKKMLEKMIEEIKKMLEKAIKKVKEMLEK-MIKEIKKMLEN---GEDSEKILKKAKEMAEKILKMVIELAEKILKKAKEMAEKILKKVKELGVDNEEVKKMLEMIEEIKKMLEKAIKKVKEMLEKMIKEIKKMLENGEDSEKILKKAKEMAEKILKMVIELAEKILKKAKEMAEKILKKVKEL-------VG |
| 9 | 1b3uA2 | 0.10 | 0.07 | 2.65 | 0.60 | HHsearch | | EDLEALVMPTLRQAAEDKSWR---VRYMVADLQKTDLVPAFQNLMK---------DCEAEVRAAASHKVKEFCENLS------------------A-DCRENV--IMSQILPCIKELVSDANQHVKS---ALASVIMGLSPIL--------GKDNTIEHLLPLFLAQLKDECPEVRLNIISNLDCVNEVI-------- |
| 10 | 3s4wA3 | 0.09 | 0.09 | 3.31 | 0.36 | CEthreader | | SQEDASFCKSLMNLFFSLHVLYKSPVTLLRDLSQ-DIHGQLGDIDQDVEIEKTDHFAVVNLRTAAPTVCLLVLSQAEKVLEEVDWLIAKIKGSAPTLLIEKAIVMQLGTLVTFFHELVQTALCVDTLGLSKIYSTLTAFVKYYLIPNTVEKLVKLSGSHLTPVCYSFISYVQNKSIPNLVFAIEQYEKFLIQLSKKSK |
| (a) | ID1 is the number of template residues identical to query divided by number of aligned residues. |
| (b) | ID2 is the number of template residues identical to query divided by query sequence length. |
| (c) | Cov is equal the number of aligned template residues divided by query sequence length. |
| (d) | Norm. Zscore is the normalized Z-score of the threading alignments. A Normalized Z-score >1 means a good alignment and is highlighted in bold. |
| (e) | Download alignment lists the threading program used to identify the template, and provide the 3D structure of aligned regions of threading templates (threading[1-10].pdb.gz). |
| (f) | Template residues identical to query sequence are highlighted in color. |
|