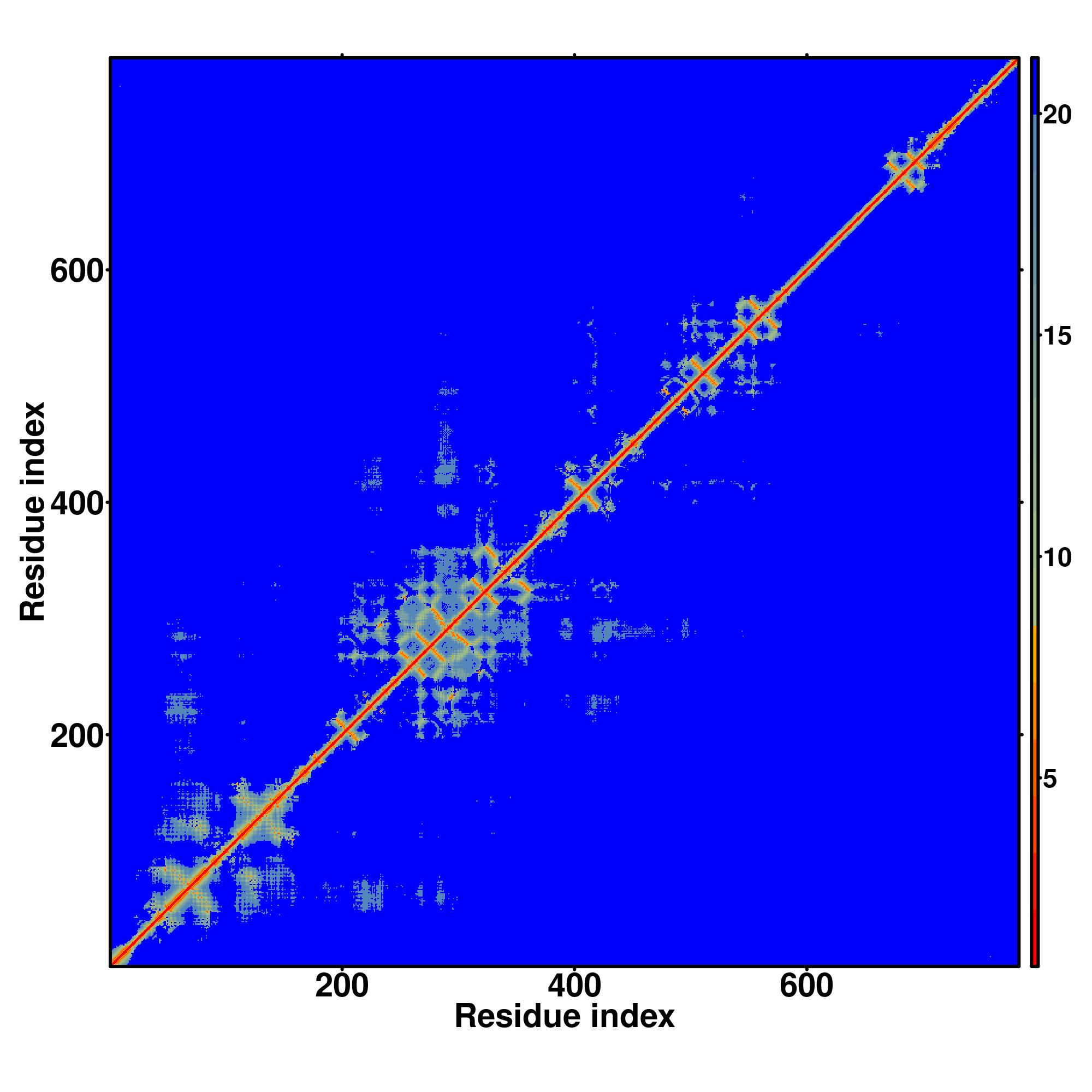
| Rank | PDB
hit | ID1 | ID2 | Cov | Norm.
Zscore | Downloadalignment | | 20 40 60 80 100 120 140 160 180 200 220 240
| | | | | | | | | | | | |
|---|
| SS
Seq | CCSSSSCCCCCCCCCCCCCSSSSSCCCCCCSSSSSSCCCCCCCCCCSCCCCCCCCCCCCCCCCCCCCCCSSSCCCCCCCCSSSSSSCCCCCCCCCCCSSSSSCCCCSSSSSSSCCCCCSSSSSSSSCCCCCSSCCCCCCCCCCCSSSSSSSCCCCCSSSSSCCCCCCCCCCCSSSSSSCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCC
MWRCSFDRGDPSSGTGRGRLFCGDVGQNKFEEVDVVERGGNYGWRAREGFECYDRSLCANTSLNDLLPIFAYPHTVGKSVTGGYVYRGCEYPNLNGLYIFGDFMSGRLMSLQENPGTGQWQYSEICMGHGQTCEFPGLINNYYPYIISFGEDEAGELYFMSTGEPSATAPRGVVYKIIDASRRAPPGKCQIQPAQVKIRSRLIPFVPKEKFIPKTRSTPRPTARAPTRAPRRGRPTAAPPAPTPRPARPTQ |
|---|
| 1 | 1cruB | 0.16 | 0.10 | 3.43 | 0.83 | DEthreader | | NPQGLAFTP-------NGKLLQSEQGPNSDDEINLIVKGGNYGWPNVAGYSYAYTKESEWTGKNFVPPLKTLYCWPTVAPSSAYVYKGGAITGWENTLLVPSLKRGVIFRIKLDPTYSTTYDAVPMFK-----------S--NNRYRDVIASPDGVLYVLTDTAGNVNLNPGSLIKFTYK----------------------------------------------------------------------- |
| 2 | 3ho4B | 0.25 | 0.22 | 6.65 | 1.37 | SPARKS-K | | PGRCAVDRHN-------LTILCSDSNGKSSARILQIIKGKDYES---------------------EPSLLEFKPFSNGPLVGGFVYRGCQSERLYGSYVFGDR-NGNFLTLQQSPVTKQWQEKPLCLGTSGSC-----RGYFSGHILGFGEDELGEVYILSSSKSTQT-HNGKLYKIVDPKRPLP-EECRATVQPAQTRNGYCT--PTGKCCCSPGWEGKCEPACRHGGVCVRPNKCLC-KKGYLGPQCEH |
| 3 | 1cruB | 0.18 | 0.11 | 3.64 | 0.76 | MapAlign | | PQGLAFTP--------NGKLLQSEQGPNSDDEINLIVKGGNYGWPNVAGYYYVPVTKESWTGKNFVPPLKTLYTVPTVAPSSAYVYKGKAITGWENTLLVPSLKRGVIFRIKLDPTYSTTYAVPM--------------FKSNNRYRDVIASPDGVLYVLTDTAGNVTLNPGSLIKFTY------------------------------------------------------------------------ |
| 4 | 3a9gA | 0.17 | 0.10 | 3.41 | 0.62 | CEthreader | | HRNPQGIDWHRASGV----MVATEHGPVGHDEVNIILKGGNYGWPLATGKAGRGEFV----------DPVIDTGSETWAPSGASFVHGDMFPGLRGWLLIACLRGSMLAAVNFGDNMEVRKISTFF-------------KNVFGRLRDVVIDDDGGILISTSNRGSLRAGDDKILKIVSEQH--------------------------------------------------------------------- |
| 5 | 3ho4B | 0.27 | 0.23 | 7.10 | 1.20 | MUSTER | | PGRCAVD-------RHNLTILCSDSGKNRSARILQIIKGKDYESE---------------------PSLLEFKPFSNGPLVGGFVYRGCQSERLYGSYVFGDR-NGNFLTLQQSPVTKQWQEKPLCLGTSGSCR-----GYFSGHILGFGEDELGEVYILSSSKS-TQTHNGKLYKIVDPKRPLPEECRATVQPAQTLTSRNGYCTPTGKCCCSPGWEGDFCRTAKCEPACRHRPNKCLCKKGYLGPQCEH |
| 6 | 3ho4B | 0.26 | 0.22 | 6.76 | 2.56 | HHsearch | | PGRCAVDR-------HNLTILCSDSGKNRSARILQIIKGKDYES---------------------EPSLLEFKPFSNGPLVGGFVYRGCQSERLYGSYVFGDR-NGNFLTLQQSPVTKQWQEKPLCLGTSGS-----CRGYFSGHILGFGEDELGEVYILSSSKS-TQTHNGKLYKIVDPKRPL-PEECRATVQPAQTLTSECSRLCRSPGWECRTAKCEPACRHGGV---CVRPNKCLCKKGYLGPQCEH |
| 7 | 3a9gA | 0.17 | 0.10 | 3.29 | 1.28 | FFAS-3D | | -QGIDWH-------RASGVMVATEHGPVGHDEVNIILKGGNYGWPLATG----------KAGRGEFVDPVIDTGSETWAPSGASFVHGDMFPGLRGWLLIACLRGSMLAAVNFGDNMEVRKISTF-------------FKNVFGRLRDVVIDDDGGILISTSNRGSLRAGDDKILKIVSEQH--------------------------------------------------------------------- |
| 8 | 4a2lA2 | 0.11 | 0.08 | 2.76 | 0.72 | EigenThreader | | VSCIVEDK--------DKNLWIGTN----DGGLNLYNPITQR-------------------------FTSYTLQGIGSNNIAVYVDEKK------SLVYIGTHAGG-LSILHR-NSGQ----VENFNQRNS--------QLVNENVYAILPDGEGNLWLGTL---------SALVRFNP------EQRTIEKEKDGTPVVSKQHKRKASILPVSSNGIKDKTTNGLPNNVSFGRETEESDGLQSNQSVGQM |
| 9 | 3a9gA | 0.17 | 0.10 | 3.39 | 2.13 | CNFpred | | PQGIDWHR-------ASGVMVATEHGPVGHDEVNIILKGGNYGWPLATGKAGR---------GEFVDPVIDTGS-ETWAPSGASFVHGDMFPGLRGWLLIACLRGSMLAAVNFGD-NMEVRKISTFFK------------NVFGRLRDVVIDDDGGILISTSNRGSLRAGDDKILKIVSE----------------------------------------------------------------------- |
| 10 | 3a9gA | 0.15 | 0.09 | 3.08 | 0.83 | DEthreader | | NPQGIDWHRA-----S-GVMVATEHGPVGHDEVNIILKGGNYGWPLATGK-A-----GR--GE-FVDPVIDTGSE-TWAPSGASFVHGDMFPGLRGWLLIACLRGSMLAAVNFGDN-MEVRKISTFFK-----------NV-FGRLRDVVIDDDGGILISTSNRGRGSLGDDKILKIVSEQ-H-------------------------------------------------------------------- |
| (a) | ID1 is the number of template residues identical to query divided by number of aligned residues. |
| (b) | ID2 is the number of template residues identical to query divided by query sequence length. |
| (c) | Cov is equal the number of aligned template residues divided by query sequence length. |
| (d) | Norm. Zscore is the normalized Z-score of the threading alignments. A Normalized Z-score >1 means a good alignment and is highlighted in bold. |
| (e) | Download alignment lists the threading program used to identify the template, and provide the 3D structure of aligned regions of threading templates (threading[1-10].pdb.gz). |
| (f) | Template residues identical to query sequence are highlighted in color. |
|