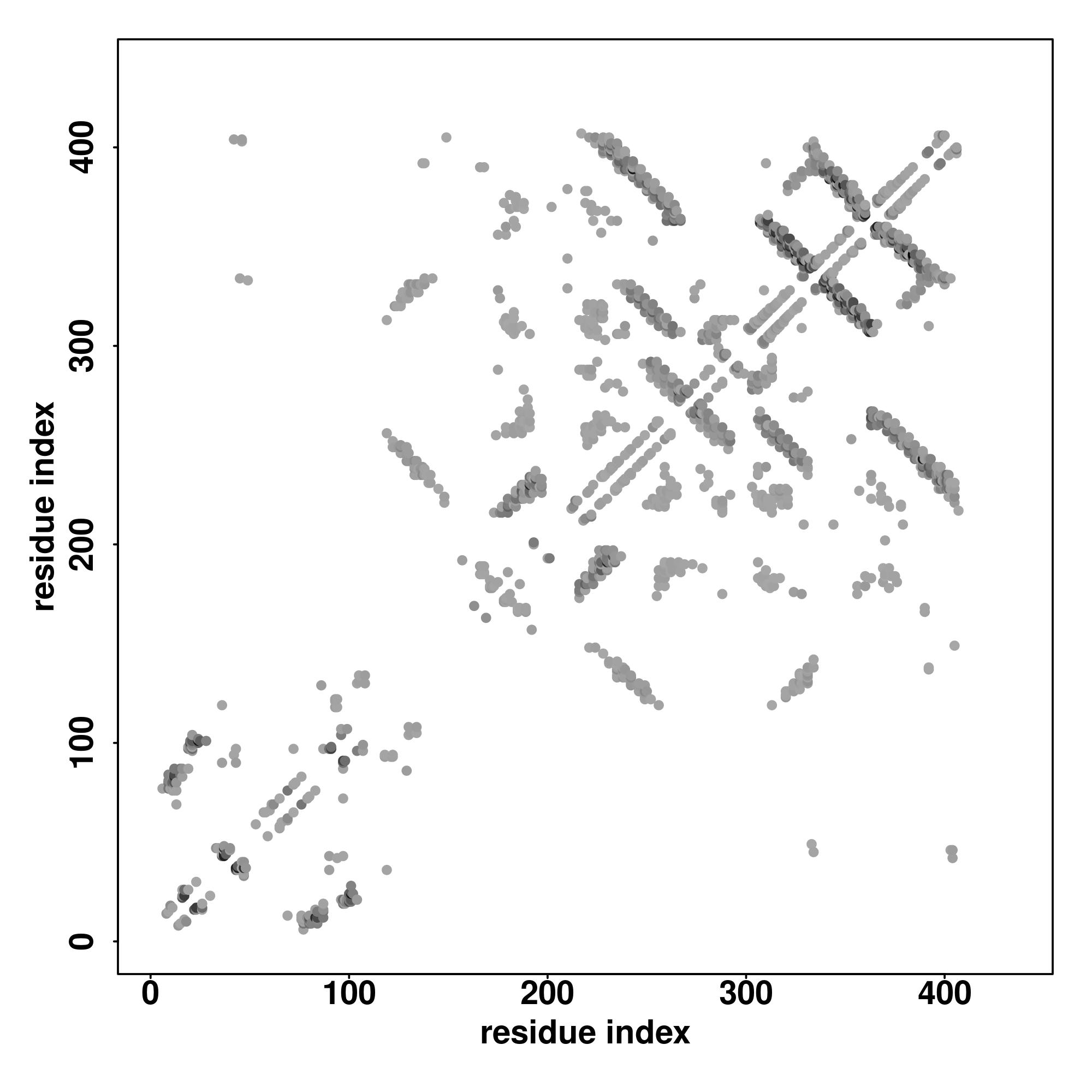
| Rank | PDB
hit | ID1 | ID2 | Cov | Norm.
Zscore | Downloadalignment | | 20 40 60 80 100 120 140 160 180 200 220 240 260
| | | | | | | | | | | | | |
|---|
| SS
Seq | CHHHHHHHHHHHHHHHHHHHHHHHHCCCCCCHHHHHCCCCCCCCCCCCCCCSSHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHCCCCCHHHHHHHHHHHHHHHHHHHHHHCCCCCCCCCHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHCCHHHHHHHHHHHHHHHHHHHHHHHCCHHHHCCHHHHHHHHHHHHHHHHHHHHHHHHHHHCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCC
SRMMQLVSSGVENALTKSELLVEQYLPLTEEELEKEAKKVEGFDLVQKPSYYVRLGSLSTKLHSRAYQQALSRVKEAKQKSQQTISQLHSTVHLIEFARKNVYSANQKIQDAQDKLYLSWVEWKRSIGYDDTDESHCAEHIESRTLAIARNLTQQLQTTCHTLLSNIQGVPQNIQDQAKHMGVMAGDIYSVFRNAASFKEVSDSLLTSSKGQLQKMKESLDDVMDYLVNNTPLNWLVGPFYPQLTESQNAQDQGAEMDKSSQETQRSEHKTH |
|---|
| 1 | 1sziA | 0.38 | 0.26 | 7.84 | 1.77 | SPARKS-K | | -----------------------NRLPLTEAELALIATPPEDSDQRQEQNYFVRLGSLSERLRNHAYEHSLGKLQNARQKAQETLQQLTSVLGLMESVKQ-----------------------------AKPE------QVEARALSMFRDITQQLQSMCVALGASIQGLPSHVREQAQQARSQVNDLQATFSGIHSFQDLSAGVLAQTRERIARAREALDNTVEYVAQNTPAMWLVGPFAPGITE-------------------------- |
| 2 | 1sziA | 0.32 | 0.23 | 6.97 | 1.13 | MapAlign | | ---------NRLPLTEAELALIATPPEDSDMASLQQQRQ--------EQNYFVRLGSLSERLRNHAYEHSLGKLQNARQKAQETLQQLTSVLGLMESVK-----------------------------------QAKPEQVEARALSMFRDITQQLQSMCVALGASIQGLPSHVREQAQQARSQVNDLQATFSGIHSFQDLSAGVLAQTRERIARAREALDNTVEYVAQNTPAMWLVGPFAPGITE-------------------------- |
| 3 | 1sziA | 0.39 | 0.27 | 7.94 | 1.35 | MUSTER | | -----------------------NRLPLTEAELALIATPPEDSDMAQEQNYFVRLGSLSERLRNHAYEHSLGKLQNARQKAQETLQQLTSVLGLMESVKQA-----------------------------------KPEQVEARALSMFRDITQQLQSMCVALGASIQGLPSHVREQAQQARSQVNDLQATFSGIHSFQDLSAGVLAQTRERIARAREALDNTVEYVAQNTPAMWLVGPFAPGITE-------------------------- |
| 4 | 1sziA | 0.39 | 0.27 | 7.94 | 7.74 | HHsearch | | -----------------------NRLPLTEAELALIATPPEDSDMRQEQNYFVRLGSLSERLRNHAYEHSLGKLQNARQKAQETLQQLTSVLGLMESVKQA--------------------------------K---PEQVEARALSMFRDITQQLQSMCVALGASIQGLPSHVREQAQQARSQVNDLQATFSGIHSFQDLSAGVLAQTRERIARAREALDNTVEYVAQNTPAMWLVGPFAPGITE-------------------------- |
| 5 | 1sziA | 0.39 | 0.27 | 7.94 | 2.20 | FFAS-3D | | -----------------------NRLPLTEAELALIATPPEDSDMAQEQNYFVRLGSLSERLRNHAYEHSLGKLQNARQKAQETLQQLTSVLGLMESVKQAKP-----------------------------------EQVEARALSMFRDITQQLQSMCVALGASIQGLPSHVREQAQQARSQVNDLQATFSGIHSFQDLSAGVLAQTRERIARAREALDNTVEYVAQNTPAMWLVGPFAPGITE-------------------------- |
| 6 | 1sziA | 0.39 | 0.26 | 7.83 | 1.27 | CNFpred | | ------------------------RLPLTEAELALIATPPEDSDMAS-QNYFVRLGSLSERLRNHAYEHSLGKLQNARQKAQETLQQLTSVLGLMESVKQ-----------------------------------AKPEQVEARALSMFRDITQQLQSMCVALGASIQGLPSHVREQAQQARSQVNDLQATFSGIHSFQDLSAGVLAQTRERIARAREALDNTVEYVAQNTPAMWLVGPFAPGITE-------------------------- |
| 7 | 3i9yA | 0.12 | 0.08 | 3.01 | 1.03 | FFAS-3D | | SAMIEAVSELSTRIISSVQMLSN---AQNEQERKEAGR---------------VLFEQLESLLTHIKEKLLDALESNVQNVINNLAELGVTVERKLWLAKEIDTRVEEMRLLSEELEQL---------TRTLDLTERLHELHLLAFKMIQQIQTAFENNLKIMKRRVLAVEPTRSKQMSQLLTELQVVFTILLQQYENNEQSQQLMQKTLELFSELNSTVNKLVD----------------------------------------------- |
| 8 | 4ui9I | 0.09 | 0.07 | 2.50 | 0.83 | DEthreader | | -------TSFYEDESNLLLIIKLLGLNILVLLYCLSLTLLYSFLP-------EVTRMAR--KF-THISALLQYINLSLTCMCEAWEEILMQMDSRLTKFVKAELQTLLMN-QL------------------------TVKGLKKLGQSIESSYSSIQKLVISLQSGSELITGSFILKANELLQVIDSSMKNFKAFFRLYAVLTEHFFNVERVGQYLKLSLHFVKRRMENIIDQCL---P--------------------------------- |
| 9 | 6w2rA | 0.14 | 0.11 | 3.67 | 0.81 | SPARKS-K | | -GTTEDERRELEKVARKAIEAAREG--NTDEVREQLQRALEIARESGT-----------KTAVKLALDVALRVAQEAAKRGNK--DAIDEAAEVVVRIAEES-NNSDALEQALRVLEEIAKAVLKSEKTE--------------DAKKAVKLVQEAYKAAQRAIEAAKGTPDVIKLAIKLAKLAARAALEVIKRPKS---------EEVNEALKKIVKAIQEAVESLREAS----------------GDPEKREKARERVREAVERAEEV-- |
| 10 | 2kbbA | 0.06 | 0.03 | 1.43 | 0.89 | MapAlign | | --GIDPFTAPGQLECETAIAALN--------------------------------------SCLRDLDQASLSQEALHTQMLTAVQEISHLIEPLASAAR------------------------------------AEASQLGHKVSQMAQYFEPLTLAAVGAASKT-----LSHPQQMALLDQTKTLAESALYTAGGNAL-EEAVQMMTEAVEDLTTTLNEAASAAG-------------------------------------------- |
| (a) | ID1 is the number of template residues identical to query divided by number of aligned residues. |
| (b) | ID2 is the number of template residues identical to query divided by query sequence length. |
| (c) | Cov is equal the number of aligned template residues divided by query sequence length. |
| (d) | Norm. Zscore is the normalized Z-score of the threading alignments. A Normalized Z-score >1 means a good alignment and is highlighted in bold. |
| (e) | Download alignment lists the threading program used to identify the template, and provide the 3D structure of aligned regions of threading templates (threading[1-10].pdb.gz). |
| (f) | Template residues identical to query sequence are highlighted in color. |
|