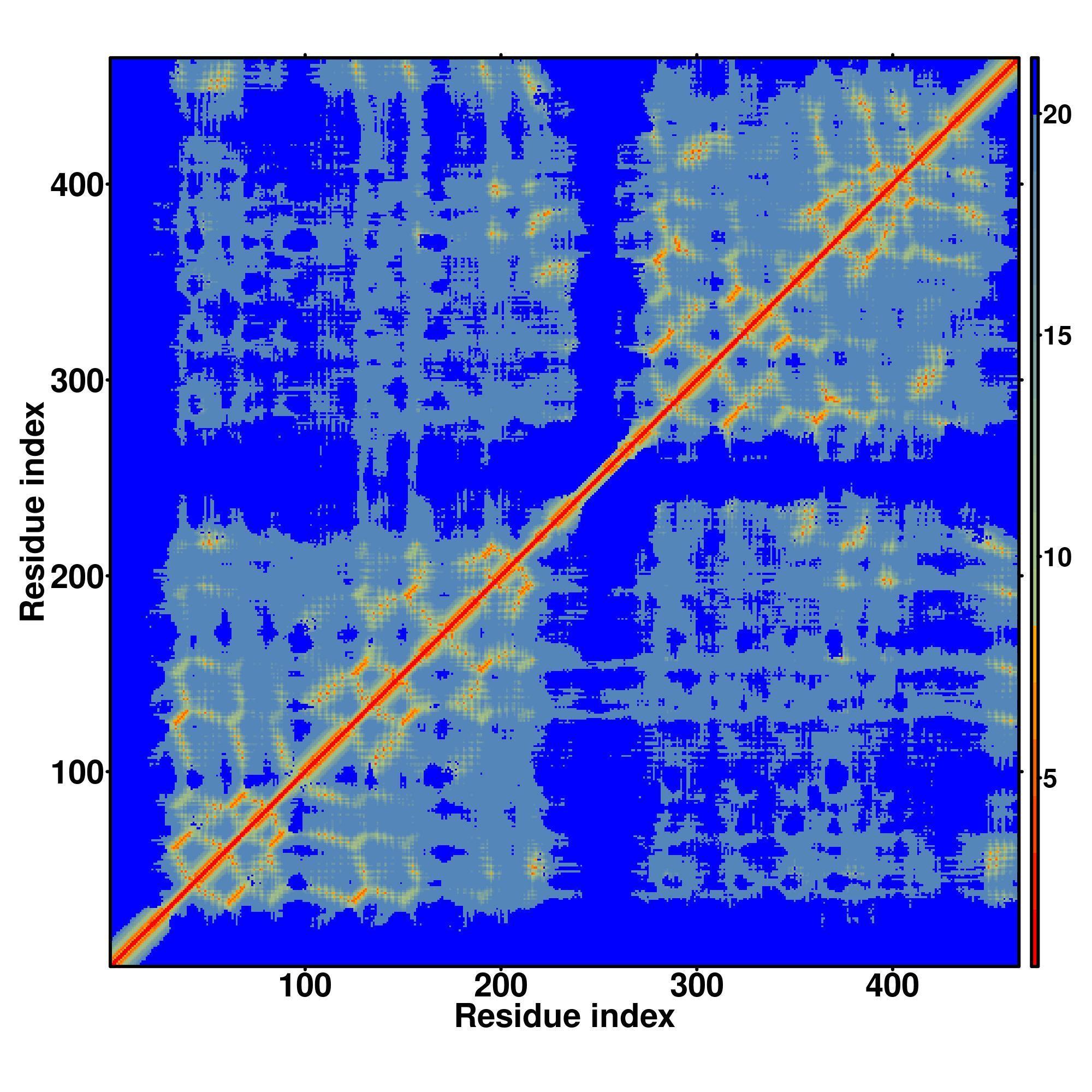
| Rank | PDB
hit | ID1 | ID2 | Cov | Norm.
Zscore | Downloadalignment | | 20 40 60 80 100 120 140 160 180 200
| | | | | | | | | | |
|---|
| SS
Seq | CHHHHHHHHHHHHHHHHHHHCCCCCCCCCCCCCSSSSSSSCCCCCCHHHHHHHHHHHHCCCSSSSSSSCCCCCCHHHHCCCCSSSSSCCCCCCCCCCHHHHHHHHHHHHHHHHHHHHHHHHCCCCSSSSSCCCCCHHHHHHHHHHHHHCCCSSSSCCCCCHHHHHHCCCCCCHHHHHHHHHHHHHHHCCCSSSHHHHHHHHHHHHHCCCCCSSC
MAASCLVLLALCLLLPLLLLGGWKRWRRGRAARHVVAVVLGDVGRSPRMQYHALSLAMHGFSVTLLGFCNSKPHDELLQNNRIQIVGLTELQSLAVGPRVFQYGVKVVLQAMYLLWKLMWREPGAYIFLQNPPGLPSIAVCWFVGCLCGSKLVIDWHNYGYSIMGLVHGPNHPLVLLAKWYEKFFGRLSHLNLCVTNAMREDLADNWHIRAVTV |
|---|
| 1 | 4rbnA | 0.07 | 0.06 | 2.36 | 1.17 | DEthreader | | --------------------------VHAIMISRLLILSPGYFGTVVYILDQVRALEKEHPKILIVTRLITTCQLEKVSGCNTWILRVPFIPHWIS--RF-EIWPHLEIFAGDVEREALAGGHPDLIIGNYS-D--GNLVATLLSRRLGVTQCNIAHALEKTKYLHSDIYWQEHFSCQYTADLLAMNSADFIVTSTYQEIAGYESQIDDKFNIS |
| 2 | 6kihA1 | 0.09 | 0.07 | 2.71 | 1.05 | SPARKS-K | | -------------------------------RQPIALISVHGDPQNIYVRQLGEALAAAGWHVDMFTRKTDPNDPDVIHSPHCRTIRLQAGPLTYIPR---EKLFETLPKFVEAFKAYHAKYGYPLIHTNYW---LSGWVGWQLRQQFNFQWLHTYHSLGVVKYQVASEQAQRD-ETRLMVEKAILENADCVIVTSPQEEAYLRRWVSKAGQTR |
| 3 | 4rbnA | 0.07 | 0.07 | 2.74 | 0.66 | MapAlign | | -NRVAETMNMLMDILPSPSALEEFLAC-IPMISRLLILSPHGYFGQVYILDQVRALEKVEPKILIVTRLICNQRLEKVSGCNTWILRVPFRKHIPHWISRFEIWPHLEIFAGDVEREALAELHPDLIIGNY---SDGNLVATLLSRRLGVTQCNIAHALEKTKSDIYWNEDKYHFSCQYTADLLAMNSADFIVTSTYQEIGQYESYGILKFNIV |
| 4 | 6d9tA1 | 0.10 | 0.08 | 2.93 | 0.51 | CEthreader | | -----------------------------QGHMKIGITCYPSMGSGIIATELGIKLAERGHEVHFITSNIPFRIRKP--LPNMIFHQVEV-----NQYAVFQYPPYDITLSTKIAEVIKE-YDLDLLHMHYA--VPHAICGILAREMSDIKIMTTLHGTDITVLGYD--------HSLQGAIKFGIEKSDIVTSVSKSLAQETHEIIETNKEII |
| 5 | 6d9tA1 | 0.09 | 0.07 | 2.67 | 0.84 | MUSTER | | -----------------------------QGHMKIGITCYPSMGSGIIATELGIKLAERGHEVHFITSNIPFRIRK--PLPNMIFHQVEVNQYAVF------QYPPYDITLSTKIAEVIKEYDLDLLHMHYAV--PHAICGILAREMSDIKIMTTLHGTDITVLGYDHS--------LQGAIKFGIEKSDIVTSVSKSLAQETHEIIETNKEII |
| 6 | 3c4qA1 | 0.13 | 0.10 | 3.42 | 1.33 | HHsearch | | --------------------------------MRVAMISMHDSGGMNVYLSTATELAKQGIEVDIYTRATRPSQGIVRVAENLRVINIAAGPYESELPTQLAA-------FTGGMLSFTRKVTYDLIHSHYW---LSGQVGWLLRDLWRIPLIHTAHTLAAVDTP------ES--EARRICEQQLVDNADVLAVNTQEEMQDLMHHYDADISVV |
| 7 | 6kihA1 | 0.10 | 0.08 | 2.96 | 1.21 | FFAS-3D | | -------------------------------RQPIALISVHSAGGNIYVRQLGEALAAAGWHVDMFTRKTDPNDPDVIEHPHCRTIRLQAGPLTYIPREKL---FETLPKFVEAFKAYHAKYGYPLIHTNY---WLSGWVGWQLRQQFNFQWLHTYHSLGVVKYQVASEQAQ-RDETRLMVEKAILENADCVIVTSPQEEAYLRR-WVSKARLI |
| 8 | 3s27A3 | 0.09 | 0.08 | 3.19 | 0.60 | EigenThreader | | NAERVLDIRLLLDLLEAPDPCTLETFLGRVPVFNVVILSPHGYFAQDYILDQVRALEIEKPRILILRLLPDAYDS-----EYCDILRVPKWISRFEV------WPYLETYTEDAAVELSKELNPDLIIGNYSD---GNLVASLLAHKLGVTQCTIAHALEKTKYPDSDIYWKKLDDKYHSCQFTADIFADFIITSTFQEIAGVF---DPKFNIS |
| 9 | 3c48A | 0.10 | 0.08 | 3.08 | 1.03 | CNFpred | | ------------------------------SHMRVAMISMHTSPLNVYILSTATELAKQGIEVDIYTRATRSQGEIVRVAENLRVINIAAGPYEGLSKELPTQLAAFTGGMLSFTRRE--KVTYDLIHSHY---WLSGQVGWLLRDLWRIPLIHTAHTLAAVKNS------TPESEARRICEQQLVDNADVLAVNTQEEMQDLMHHYDADPDRI |
| 10 | 4rbnA3 | 0.07 | 0.06 | 2.36 | 1.17 | DEthreader | | --------------------------VHAIMISRLLILSPGYFGTVVYILDQVRALEKEHPKILIVTRLITTCQLEKVSGCNTWILRVPFIPHWIS--RF-EIWPHLEIFAGDVEREALAGGHPDLIIGNYS-D--GNLVATLLSRRLGVTQCNIAHALEKTKYLHSDIYWQEHFSCQYTADLLAMNSADFIVTSTYQEIAGYESQIDDKFNIS |
| (a) | ID1 is the number of template residues identical to query divided by number of aligned residues. |
| (b) | ID2 is the number of template residues identical to query divided by query sequence length. |
| (c) | Cov is equal the number of aligned template residues divided by query sequence length. |
| (d) | Norm. Zscore is the normalized Z-score of the threading alignments. A Normalized Z-score >1 means a good alignment and is highlighted in bold. |
| (e) | Download alignment lists the threading program used to identify the template, and provide the 3D structure of aligned regions of threading templates (threading[1-10].pdb.gz). |
| (f) | Template residues identical to query sequence are highlighted in color. |
|