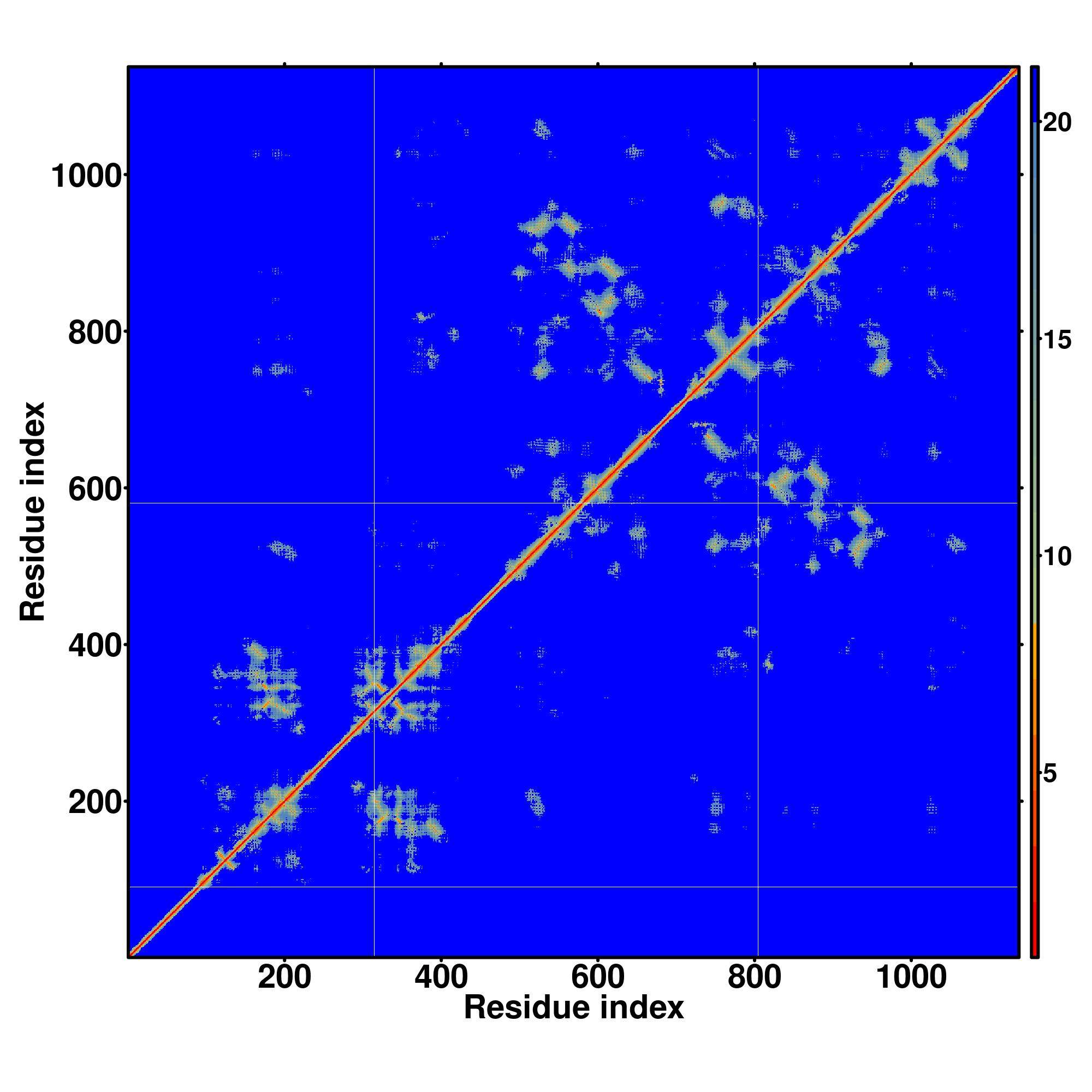
| Rank | PDB
hit | ID1 | ID2 | Cov | Norm.
Zscore | Downloadalignment | | 20 40 60 80 100 120 140 160 180 200
| | | | | | | | | | |
|---|
| SS
Seq | CCHHHHHHHHHHHHHHHHHHHHHHCCCCCCCCHHHHHHHHHHHHHHHHHHHHHHCCCCCCCCCCSSSSHHHHHHHHHHHHHHHHHHHCCHHHHHHCHHHHHHHHHHHHHHHHCCCCCHHHHHCCCCCCCCCCHHHHHHHHHCCCCCCCCCCCCCCCCCHHHHHHHHHHHHHHHHHHHHHHCCHHHHHHHHHHHHHHHHHHHHHHCCCHHHHHHHCCCCC
FTEEGFSTLISFIFIYDAIKKMIGAFKYYIPDLALMSFILFFGTYSMTLTLKKFKFSRYFPTKVRALVADFSIVFSILMFCGIDACFGLETPKLHVPLPVLYGVFLYMGVASLNGIQMGTGGSEFKIQKKLTPFWERCKLFLMPAKHQPDHAFLRHVPLRRIHLFTLVQILCLAVLWILKSTVAAIIFPVMILGLIIVRRLLDFIFSQHDLAWIDNILP |
|---|
| 1 | 6caaA | 0.62 | 0.57 | 16.20 | 1.17 | DEthreader | | RTEEGFSSLISFIFIYDAFKKMIKLADYYPIDITLMSFILFLGTYTSSMALKKFK-TSPFPTTARKLISDFAIILSILIFCVIDALVGVDTKLLKIPMPVLYGVFLYMGVASL-NG--------------V-QFMDRLKLLLMPLKHQPDFIYLRHVPLRRVHLFTFLQVLCLALLWILKSTVAAIIFPVMILALVAVRKGMDY-LFSQ--HDLSFLDD |
| 2 | 5l25A | 0.31 | 0.26 | 8.01 | 1.22 | SPARKS-K | | VAGELFGLLIAMLFMQQAIKGLVDEFRIPEFANGMFALVLSFGLLLTGLRSRKARSWRYGTGWLRSLIADYGVPLMVLVWTGVSQLAQQKVIPQAMPTSVLWGYFAFMAIESLPGNQ----------------FWERILLLFTAFKVLYHATFVETVPFKTIAMFTLFQTTYLLICFGLTWIPAGVMFPLMIMFLIPVRQYLLP--------------- |
| 3 | 4yzfA | 0.42 | 0.38 | 11.26 | 1.08 | MapAlign | | YTQEIFSFLISLIFIYETFSKLIKIFQDHPLNTALLSLVLMAGTFFFAMMLRKFKNSSYFPGKLRRVIGDFGVPISILIMVLVDFFIDTYTQLSRIPLAVLFGIFLYMGVTSLSGI----------------QLFDRILLLFKPPKYHPDVPYVKRVKTWRMHLFTGIQIICLAVLWVVKSTPASLALPFVLILTPLRRVLLPLIFRNVELQCLD---- |
| 4 | 4yzfA | 0.42 | 0.38 | 11.14 | 1.08 | CEthreader | | YTQEIFSFLISLIFIYETFSKLIKIFQDHPLNTALLSLVLMAGTFFFAMMLRKFKNSSYFPGKLRRVIGDFGVPISILIMVLVDFFIQDTYTQKRIPLAVLFGIFLYMGVTSLSGI----------------QLFDRILLLFKPPKYHPDVPYVKRVKTWRMHLFTGIQIICLAVLWVVKSTPASLALPFVLILTVPLRRVLPLIFRNVELQCLD---- |
| 5 | 5l25A | 0.31 | 0.26 | 8.01 | 0.84 | MUSTER | | VAGELFGLLIAMLFMQQAIKGLVDEFRIPEFANGMFALVLSFGLLLTGLRSRKARSWRYGTGWLRSLIADYGVPLMVLVWTGVSYIPIVYIIGAFIPTSVLWGYFAFMAIESLPGN----------------QFWERILLLFTAPSRRYHATFVETVPFKTIAMFTLFQTTYLLICFGLTWIIAGVMFPLMIMFLIPVRQYL---------------LP |
| 6 | 5l25A | 0.33 | 0.28 | 8.37 | 5.71 | HHsearch | | VAGELFGLLIAMLFMQQAIKGLVDEFRIPRFANGMFALVLSFGLLLTGLRSRKARSWRYGTGWLRSLIADYGVPLMVLVWTGVSYIPG-SIPQLAMPTSVLWGYFAFMAIESLPGNQ----------------FWERILLLFTAPSRRYHATFVETVPFKTIAMFTLFQTTYLLICFGLTWIIAGVMFPLMIMFLIPVRQLLP---------------- |
| 7 | 5l25A | 0.30 | 0.26 | 7.89 | 1.61 | FFAS-3D | | VAGELFGLLIAMLFMQQAIKGLVDEFRIPERENGMFALVLSFGLLLTGLRSRKARSWRYGTGWLRSLIADYGVPLMVLVWTQSTMVGGCVAAMPILKTSVLWGYFAFMAIESLPGN----------------QFWERILLLFTAPSRRFKVTFVETVPFKTIAMFTLFQTTYLLICFGLTIPIAGVMFPLMIMFLIPVRQYL---------------LP |
| 8 | 4yzfA | 0.37 | 0.32 | 9.63 | 1.08 | EigenThreader | | YTQEIFSFLISLIFIYETFSKLIKIFQDHPLLLSLVLMAGTFFFAMMLRKFKNSSYF---PGKLRRVIGDFGVPISILIMVLVDFFIQDTYTQKRIPLAVLFGIFLYMGVTSL-------SGI---------QLFDRILLLFKPPKYHPDVPYVKRVKTWRMHLFTGIQIICLAVLWVVKSTPASLALPFVLILTVPLRRVLLRNVELQCLD------- |
| 9 | 4yzfA | 0.27 | 0.24 | 7.42 | 1.15 | CNFpred | | FASALPALLVFILIFLESQITTLIVSSGFHLDLLLVVGMGGVAAWLSATTVRSVTHANALTVVKEQRISGLLVAVLVGLSILMEPIL------SRIPLAVLFGIFLYMGVTSLSG----------------IQLFDRILLLFKPPKYHPDVPYVKRVKTWRMHLFTGIQIICLAVLWVVKSTPASLALPFVLILTVPLRRLLPLIFRNVELQCLD---- |
| 10 | 4yzfA | 0.43 | 0.39 | 11.51 | 1.17 | DEthreader | | RTQEIFSFLISLIFIYETFSKLIKIFQDHLPNTALLSLVLMAGTFFFAMMLRKFKNSSYFPGKLRRVIGDFGVPISILIMVLVDFFIDTYTQKLSIPLAVLFGIFLYMGVTSL-SG--------------I-QLFDRILLLFKPPKYHPDVPYVKRVKTWRMHLFTGIQIICLAVLWVVKSTPASLALPFVLLTVPLRRVLLPLIFRNVELQCLD---- |
| (a) | ID1 is the number of template residues identical to query divided by number of aligned residues. |
| (b) | ID2 is the number of template residues identical to query divided by query sequence length. |
| (c) | Cov is equal the number of aligned template residues divided by query sequence length. |
| (d) | Norm. Zscore is the normalized Z-score of the threading alignments. A Normalized Z-score >1 means a good alignment and is highlighted in bold. |
| (e) | Download alignment lists the threading program used to identify the template, and provide the 3D structure of aligned regions of threading templates (threading[1-10].pdb.gz). |
| (f) | Template residues identical to query sequence are highlighted in color. |
|