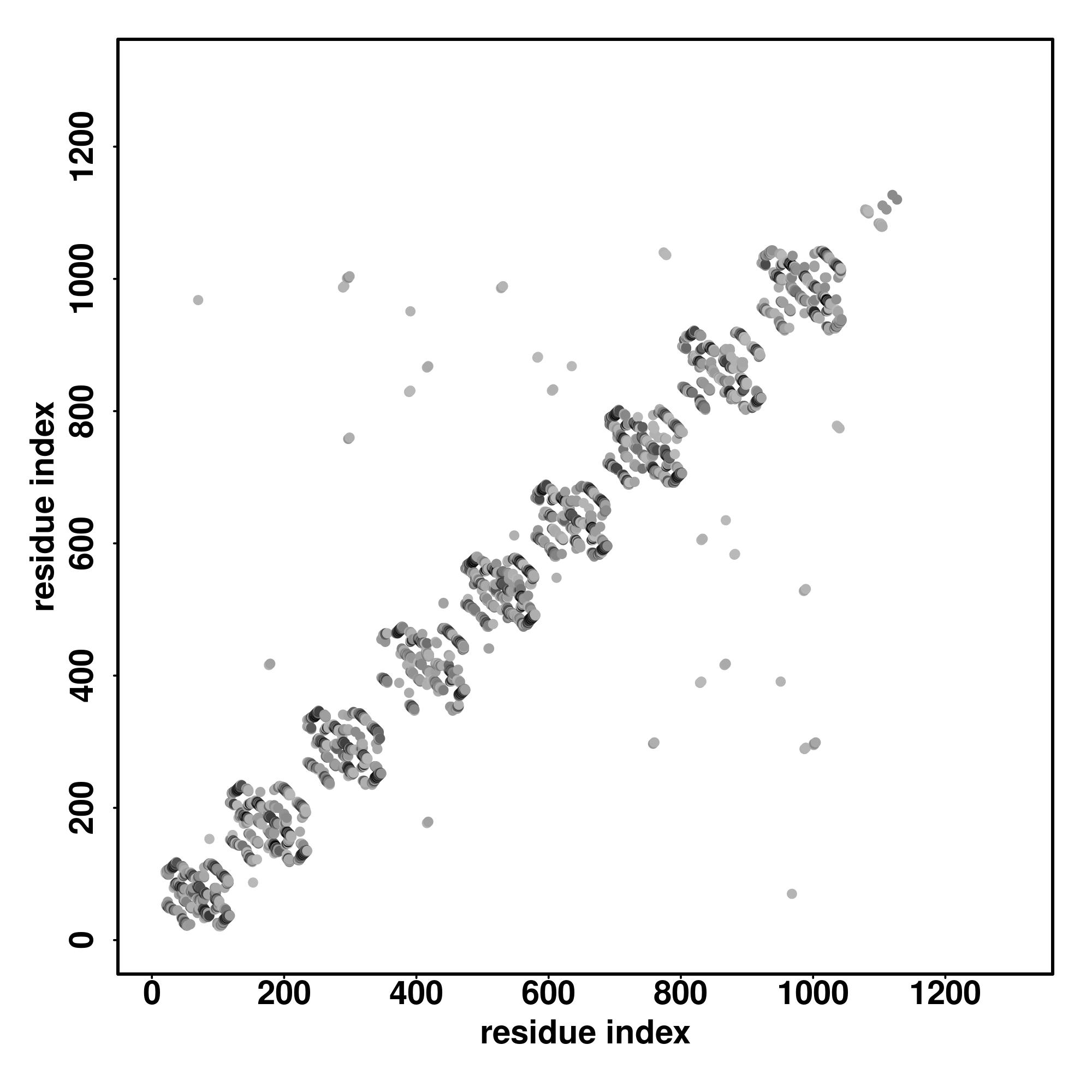
| Rank | PDB
hit | ID1 | ID2 | Cov | Norm.
Zscore | Downloadalignment | | 20 40 60 80 100 120 140 160 180 200 220 240 260
| | | | | | | | | | | | | |
|---|
| SS
Seq | CCCCCCCCCCCCCSSSSSSCCCCCCCSSSSSSSSCCCCCCCCSSSSSSSCCCCCCCCCCCCCCCCSSSSCCCCCCCCCCSSSSSCCCCCCCCCCSSSSSSSSSSCCCCCCCCSSSSSSSSSSSSCCCCCCSSSCCCCCSSSSCCCCCCCCCCCCCCSSSSSSSSSSCCCCCCCCSSSSSSSSSSCCCCCCSSSSSSSSSSSCCCCCCCSSSSSSSSSSCCCCCCCCCCCCSSSHHHHHHHHHHHHHHHHHHHHHHHHHHCCCCCCCC
VNDNAPYFLPENKTFVIIPELVLPNREVASVRARDDDSGNNGVILFSILRVDFISKDGATIPFQGVFSIFTSSEADVFAGSIQPVTSLDSTLQGTYQVTVQARDRPSLGPFLEATTTLNLFTVDQSYRSRLQFSTPKEEVGANRQAINAALTQATRTTVYIVDIQDIDSAARARPHSYLDAYFVFPNGSALTLDELSVMIRNDQDSLTQLLQLGLVVLGSQESQESDLSKQLISVIIGLGVALLLVLVIMTMAFVCVRKSYNRKLQA |
|---|
| 1 | 6c13A | 0.16 | 0.13 | 4.23 | 1.47 | SPARKS-K | | ENDHPPVFQKK-FYIGGVSEDARMFASVLRVKATDRDTGNYSAMAYRLIIPPIKEGKEG-------FVV------ETYTGLIKTAMLFHNMRRSYFKFQVIATDDY--GKGLSGKADVLVSVVNQLDMQVIVSNVPPTLVEKKIEDLTEILDRYVPGAKVVVESIGARRSLEDYSKCDLTVYAIDPQTRAIDRNELFKFLDGKLLDINKDFQPYYGEGGRILEIRTP---------------------------------------- |
| 2 | 5vh2A | 0.21 | 0.16 | 5.13 | 1.11 | MUSTER | | -NDEAPVFTQQQYNRLGLRETAGIGTSVIVVRATDKDTGDGGLVNYRILSG-----------AEGKFEID------ESTGLIVTVDYLDYETKTSYLMNVSATDGAP--PFNQGFCSVYVTLLNELDEAVQFSNASYE--------AVIMENLALGTEIVRVQAYDIDN--LNQITYRFDAYTSAQAKALFKIDAITGVITVKGLVDREKGDFYTLTVVADDGGPKVDSTVKVYITVL----------------------------- |
| 3 | 5t9tA5 | 0.32 | 0.13 | 3.96 | 1.70 | FFAS-3D | | -NDNAPVFHQAS-YLVHVAENNPPGTSIAQVSASDPDLGSNGLISYSIIAS-----DLEPRALSSFVSV------NQDSGVVFAQRAFDHEQLRSFQLTLQARDHGS--PTLSANVSMRVLVGDRN--------------------------------------------------------------------------------------------------------------------------------------------- |
| 4 | 6bxzC | 0.16 | 0.12 | 4.10 | 1.58 | CNFpred | | ENDHPPVFQK-KFYIGGVSEDARMFASVLRVKATDKDTGNYSAMAYRLIIP-------PIKEGKEGFVVET------YTGLIKTAMLFHNMRRSYFKFQVIATDDYG--KGLSGKADVLVSVVNQLDMQVIVSNVPPTLVEKKIEDLTEILDRYVPGAKVVVESIGARRHLEDYTKCDLTVYAIDPQNRAVDRNELFKFLD-INKDFQPYYGGRILEIRTPE--------------------------------------------- |
| 5 | 6c13A | 0.15 | 0.10 | 3.55 | 0.83 | DEthreader | | LENDHPPVFQKKFYIGGVSEDARMFASVLRVKATDTGNY--SAMAYRLIIPPI-------KEGKEGFVVETY------TGLIKTAMLFHNMRRSYFKFQVIATDDYG--KGLSGKADVLVSVVNQLDMQVIVSNVPP--VK--LEILDRYVQEQIPAKVVVESIGARRLED-YS-KCDLTVYAIDPQNRAIDRNELF--K--F--LDGKLLDINIEIRTP----------------------------------------------- |
| 6 | 7agfD | 0.18 | 0.14 | 4.51 | 1.23 | SPARKS-K | | INDNEPVFTQDV-FVGSVEELSAAHTLVMKINATDADEPLNSKISYRIVSLE--------PAYPPVFYLN------KDTGEIYTTVTLDREEHSSYTLTVEARDGNGEVTKPVKQAQVQIRILDVNDNIVLEGMVEENQ---------------VNVEVT--RIKVFDADEIGSDNWLANFTFASGEGGYFH-------IETDAQTNEEMKNLDFSVIVANKAAFHKSIRSKYKPTPIPIKVKV----------------------- |
| 7 | 6c13A | 0.16 | 0.12 | 4.00 | 0.55 | MapAlign | | ENDHPPVFQK-KFYIGGVSEDARMFASVLRVKATDRDT-GNYAMAYRLI-IPPI------KEGKEGFVVET------YTGLIKTAMLFHNMRRSYFKFQVIATDDYG--KGLSGKADVLVSVVNQLDMQVIVSNVPPTLVEDLTEILDRYVQEQIGAKVVVESIGARRYSLEDYSKCDLTVYAIDPTNRAIDRNELFKFLDGKLLDINKDFGGRILEIRTP---------------------------------------------- |
| 8 | 6c13A | 0.15 | 0.12 | 4.03 | 0.31 | CEthreader | | ENDHPPVF-QKKFYIGGVSEDARMFASVLRVKATDRDTGNYSAMAYRLIIPPI-------KEGKEGFVVE------TYTGLIKTAMLFHNMRRSYFKFQVIATDDYG--KGLSGKADVLVSVVNQLDMQVIVSNVPPTLVEKKIEDLTEILDRYVQAKVVVESIGARRSLEDYSKCDLTVYAIDPQTNRAIDRNELFKFLDGKLLDINKDFQPYYGEGGRILEIRTP---------------------------------------- |
| 9 | 5wjmA | 0.20 | 0.16 | 5.02 | 1.00 | MUSTER | | --DNVPTFPRDY-EGPFDVTEGQPGPRVWTFLAHDRDSGPNGQVEYSVVDG----------DPLGEFVISP------VEGVLRVRKDLDRETIAFYNLTICARDRGV--PPLSSTMLVGIRVLDINDNDPVLLNLP--------MNVTISENSPVSSFVAHVLASDADSGCNALLTFNITA----NRERAFFINATTGIVTVNRPLDRERIPE-TVSVKDNPENPRIARKDFDLLLVSLA--------------------------- |
| 10 | 6c13A | 0.16 | 0.12 | 4.10 | 0.87 | HHsearch | | ENDHPPVFQKKF-YIGGVSEDARMFASVLRVKATDRDTGNYSAMAYRLIIPP-------IKEGKEGFVV------ETYTGLIKTAMLFHNMRRSYFKFQVIATDDYG--KGLSGKADVLVSVVNQLDMQVIVSNVPPTLVEKKIEDLTEILDQIPGAKVVVESIGARRHGLEDYSKCDLTVYAIDPQTRAIDRNELFKFLDGKLLDIKDFQPYRILEIRTP---------------------------------------------- |
| (a) | ID1 is the number of template residues identical to query divided by number of aligned residues. |
| (b) | ID2 is the number of template residues identical to query divided by query sequence length. |
| (c) | Cov is equal the number of aligned template residues divided by query sequence length. |
| (d) | Norm. Zscore is the normalized Z-score of the threading alignments. A Normalized Z-score >1 means a good alignment and is highlighted in bold. |
| (e) | Download alignment lists the threading program used to identify the template, and provide the 3D structure of aligned regions of threading templates (threading[1-10].pdb.gz). |
| (f) | Template residues identical to query sequence are highlighted in color. |
|