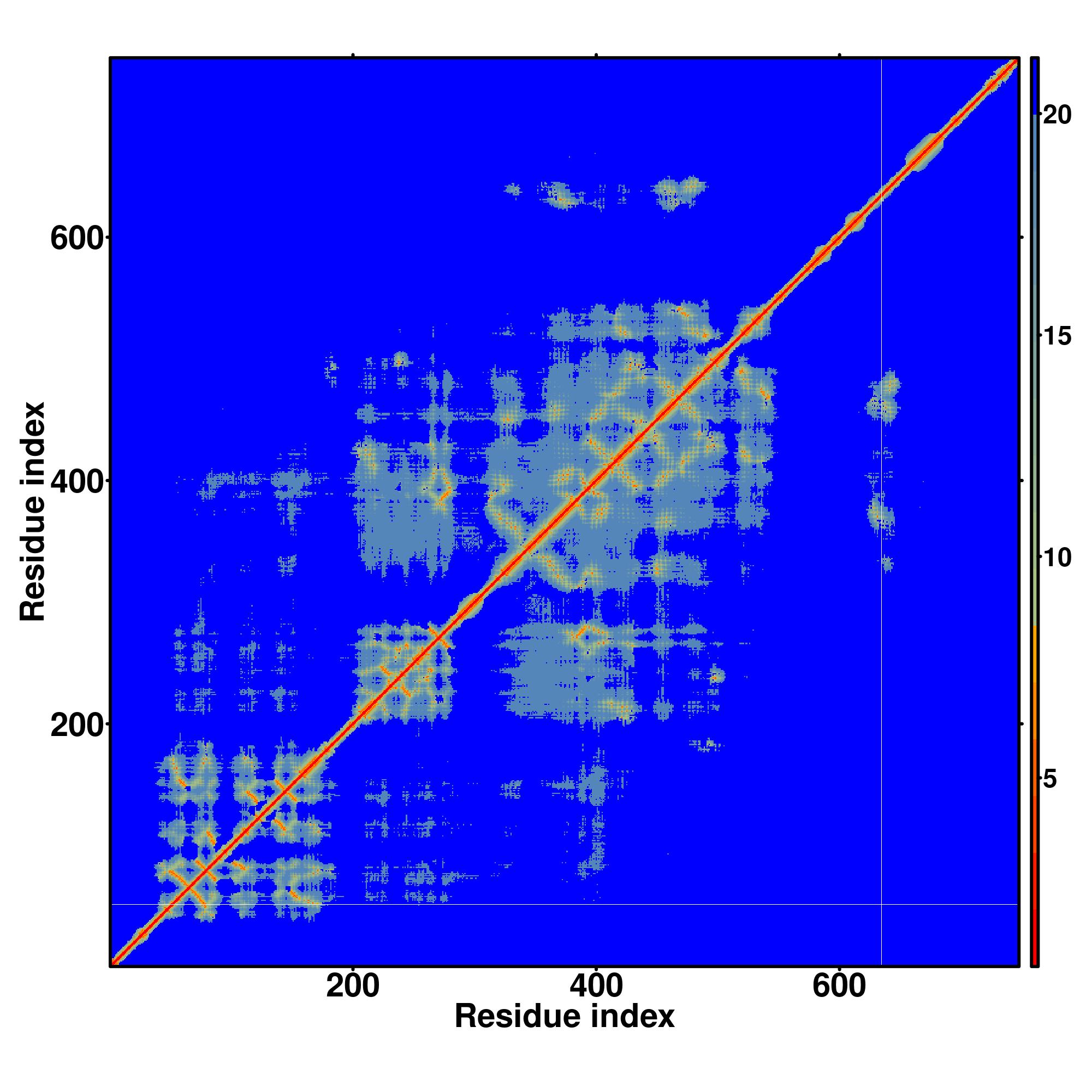
| Rank | PDB
hit | ID1 | ID2 | Cov | Norm.
Zscore | Downloadalignment | | 20 40 60 80 100 120 140 160 180 200 220 240 260
| | | | | | | | | | | | | |
|---|
| SS
Seq | CCCCCCCCCCCCCCCCCCCSSSCCCCCCCCCCCCCCCCCCCCCCCCCCCSSSSSSCCSSSSCCCCCCCCCCSSSSSSSCCSSSSSSCCCCCCCCCCCCCCCCSCCCCCCCCSSSSSSSSSCCCCCCCCCCCCCCCCCSSSSSSCCCCSSSSSSCCCCHHHHHHHHHHHHHHCCHHHHCCCCCCCCCCCCCCCCCHHHHHHHHHHCCCCCCHHHHHHCCCCCCCCSSSCSCCCCCCCCCCCCSSSSCCCCCCCCCHHHHHHHCHHCCCCCCC
MLGKGVVGGGGGTKAPKPSFVSYVRPEEIHTNEKEVTEKEVTLHLLPGEQLLCEASTVLKYVQEDSCQHGVYGRLVCTDFKIAFLGDDESALDNDETQFKNKVIGENDITLHCVDQIYGVFDEKKKTLFGQLKKYPEKLIIHCKDLRVFQFCLRYTKEEEVKRIVSGIIHHTEAQILGGGQVATLSKLLEMMEEVQSLQEKIDERHHSQQAPQAEAPCLLRNSARLSSLFPFALLQRHSSKPVLPTSGWKALGDEDDLAKREDEFVDLGDV |
|---|
| 1 | 2vdcA | 0.05 | 0.04 | 1.85 | 0.83 | DEthreader | | --PRRVVKGIEA---PQKFFKVPGNNGVSDEQELYSSRMLAHNGE-----NTVDSSLMLVPQLTDNALICNMEPWD-----GPAALAMTDGRW------VVGGMDNGLRNPPIDSRMSLKTRQLESPTHFAIGATT---LLK------ISVIS--SYRG-GGN-FE------------------ITPGM-S------MGALS-AH-LMKSDSSAIKQVAREIKVAQKGEG--GQLPGKVIARLRHGMFSATANSGCYMTAGMTNDES---- |
| 2 | 1zvrA | 0.18 | 0.13 | 4.34 | 1.97 | SPARKS-K | | ---------------------------------------MEEPPLLPGENIKDMAKDVTYICPF---TGAVRGTLTVTNYRLYFKSMERD-----------PPF-VLDASLGVINRVEKIGGA------SSRGENSYGLETVCKDIRNLRFAHKP-EGRTRRSIFENLMKYAFPVSNNPNESWRITKINERYELCDTYPALLVVPA-------NIPDEELKRVASFSWIHPSQATITRCSQPMVGVSGKRSKEDEKYLQAIMDSNAQSHKI |
| 3 | 1zvrA | 0.15 | 0.12 | 3.96 | 0.97 | MapAlign | | ----------------------------------------EEPPLLPGENIKDMAKDVTYIC---PFTGAVRGTLTVTNYRLYFKSMER-----------DPPF-VLDASLGVINRVEKIGGASSRGEN------SYGLETVCKDIRNLRFAHK-PEGRTRRSIFENLMKYAFPVSNNLPLFNESWRITKINERYELCDTYPALLVVPANIPDEELKRVASFPVLSWIHPESQATITRCSQPMVGVSGKRSKEDEKYLQAIMDSNAQSHKI |
| 4 | 1zvrA | 0.17 | 0.13 | 4.36 | 0.79 | CEthreader | | ---------------------------------------MEEPPLLPGENIKDMAKDVTYICPF---TGAVRGTLTVTNYRLYFKSMERDP------------PFVLDASLGVINRVEKIGGA------SSRGENSYGLETVCKDIRNLRFAHKPE-GRTRRSIFENLMKYAFPVSNNLPLFAFETKINERYELCDTYPALLVVPANIPDEKRVASFRSRGRIPVLSWIHPEQATITRCSQPMVGVSGKRSKEDEKYLQAIMDSNAQSHKI |
| 5 | 1zvrA | 0.18 | 0.14 | 4.44 | 1.04 | MUSTER | | ---------------------------------------MEEPPLLPGENIKDMAKDVTYIC---PFTGAVRGTLTVTNYRLYFKSMERDPP------------FVLDASLGVINRVEKIGGASS------RGENSYGLETVCKDIRNLRFAHKPEGR-TRRSIFENLMKYAFPVSNNPNESWRITKINERYELCDTYPALLVVP-------ANIPDEELKRVAVLSWIHPESATITRCSQPMVGVSGKRSKEDEKYLQAIMDSNAQSHKI |
| 6 | 1zvrA | 0.19 | 0.14 | 4.63 | 5.53 | HHsearch | | ---------------------------------------MEEPPLLPGENIKDMAKDVTYICPF---TGAVRGTLTVTNYRLYFKSMERDPP------------FVLDASLGVINRVEKIGGAS------SRGENSYGLETVCKDIRNLRFAHKPEGR-TRRSIFENLMKYAFPVSNNGNESWRITKINERYELCDTYPALLVV-------PANIPDEELKRVPVLSWIHPEATIT-RCSQPMVGVSGKRSKEDEKYLQAIMDSNAQSHKI |
| 7 | 1zvrA1 | 0.19 | 0.08 | 2.49 | 1.46 | FFAS-3D | | -----------------------------------------EPPLLPGENIKDMAKDVTYICP---FTGAVRGTLTVTNYRLYFKSMERDPP------------FVLDASLGVINRVEKIGGAS------SRGENSYGLETVCKDIRNLRFAHK-PEGRTRRSIFENLMKYA--------------------------------------------------------------------------------------------------- |
| 8 | 4gzuA | 0.07 | 0.05 | 2.18 | 0.87 | EigenThreader | | SSAHLKGDHQRIAHYSPGH-----RDNLQKLTELQRDLVGVENLIAPGREFIREGC--LHKLT---KKGLQQRMFFLFSDMLLYTSKS---VTGA-----SHFRIRGFLPLRGMLVEES-----------NEWSVLHCFTIYAAQKTIVVAAS---TRLEKEKWMQDLNAAIQAAHVCWYRNTSVSRADHSAAVENQNGFCLFFYKTHQDDYPLLGY------SVSLPRKDYFRA-------------ESKYTFERWMDVIKR--AS---- |
| 9 | 1zsqA | 0.18 | 0.13 | 4.33 | 2.49 | CNFpred | | ---------------------------------------MEEPPLLPGENIKDMAKDVTYICPF---TGAVRGTLTVTNYRLYFKSMERDP------------PFVLDASLGVINRVEKIGGASSRGE------NSYGLETVCKDIRNLRFAHKPEG-RTRRSIFENLMKYAFPVSN-PNESWRITKINERYELCDTYPALLVVPAN-------IPDEELK-IPVLSWIHPEQATITRCSQPMVGVSGKRSKEDEKYLQAIMDSNAQSHKI |
| 10 | 7czeA | 0.04 | 0.03 | 1.36 | 0.67 | DEthreader | | -----------------------LASMLNRYKLIYKT-----EPVDIPMSLELFHLSYVALSINGDKFQYTGAMTSK-----FLMGSLIVTTFVHNFYFPLTMFEVSAFSFFELTV-----------------------LKDIIGICYGATV--------K--GM-QSYGLER-LA--A--MLMATV-KME--ELG-H-------------L--TTEKQEYALRLATVGYPAGSGLIGATSLLSAYNRHLMRETLFIGSHVVLRELRLNVT |
| (a) | ID1 is the number of template residues identical to query divided by number of aligned residues. |
| (b) | ID2 is the number of template residues identical to query divided by query sequence length. |
| (c) | Cov is equal the number of aligned template residues divided by query sequence length. |
| (d) | Norm. Zscore is the normalized Z-score of the threading alignments. A Normalized Z-score >1 means a good alignment and is highlighted in bold. |
| (e) | Download alignment lists the threading program used to identify the template, and provide the 3D structure of aligned regions of threading templates (threading[1-10].pdb.gz). |
| (f) | Template residues identical to query sequence are highlighted in color. |
|