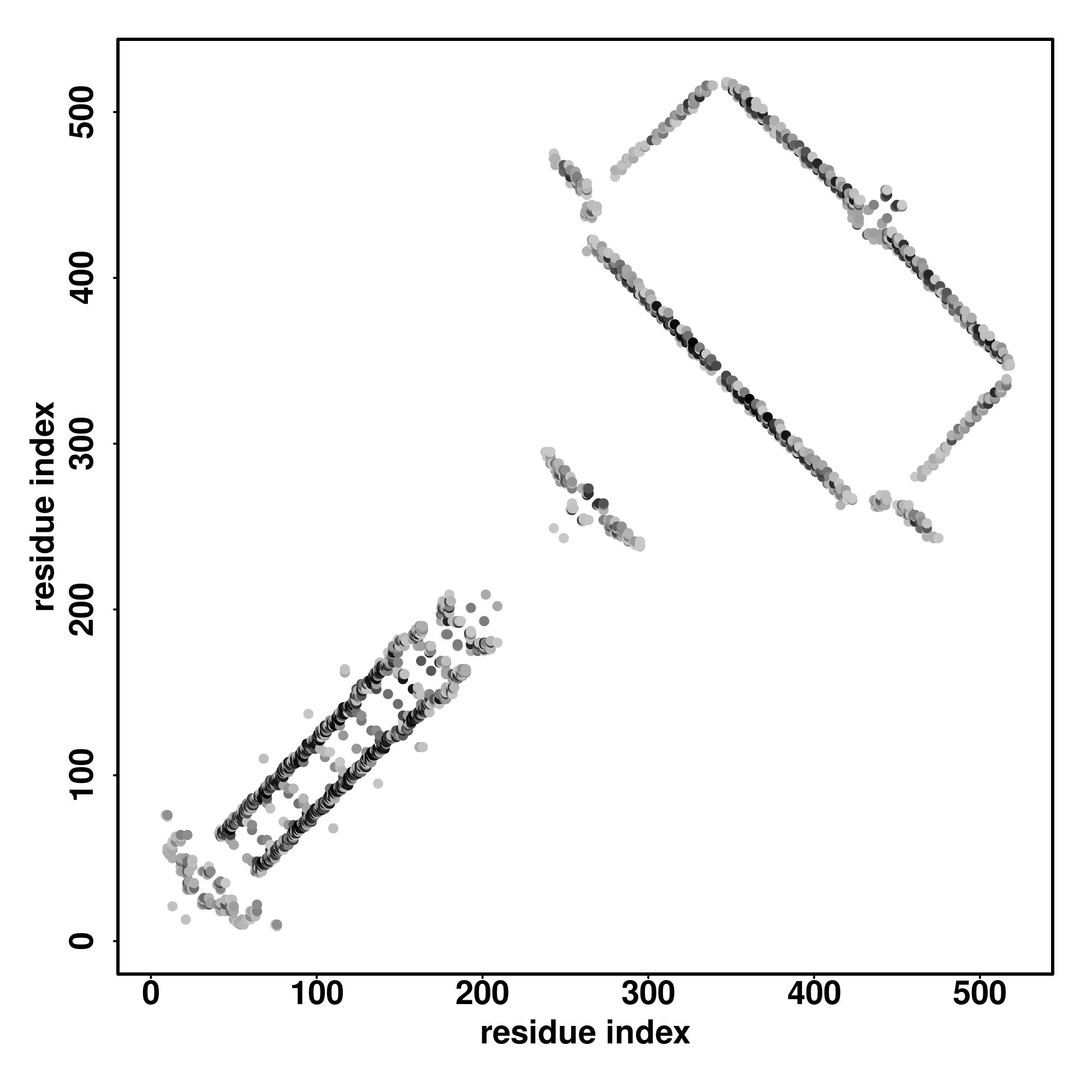
| Rank | PDB
hit | ID1 | ID2 | Cov | Norm.
Zscore | Downloadalignment | | 20 40 60 80 100 120 140 160 180 200 220 240 260
| | | | | | | | | | | | | |
|---|
| SS
Seq | CCCCCHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHCCCCHHHHHHHHHCCCHHHHHHHCCCHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHCCCCCCCCHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHCCC
YRRIDDHTKKLAEAKHQYSIDELKHQENLMQAQLEDEQAQREELEKHKTAFVEHLNGSFLFDSMYAEDSEGNNLSYLPGVGELLETYKDKFVIICVNIFEYGLKQQEKRKTELDTFSECVREAIQENQEQGKTLEMQLVEQLEETINMFERNIVDMVGLFIENVQSLMAQCRDLENHHHEKLLEISISTLEKIVEGDLDEDLPNDLRALFVDKDTIVNAVGASHDIHLLKIDNREDELVTRINSWCTRLIDRIHKDEIMRNRK |
|---|
| 1 | 3i9wA | 0.07 | 0.07 | 2.76 | 1.24 | FFAS-3D | | ------SEASAWELFAAQNLTSADNEKMWQAQGRMLTAQSLKINALLQALREQGFDTTAIEQEI------SRSLRQQGELVGQRLQLRQQQQQLSQQIVAAADEIARLAQGQANNATTSAGATQAGIRQAAESALDRLIDIYVNQMNELRLSALRVQQMVMNGLEQIQKNAPTLEKQLNNAV-----KILQRRQIRIEDPGVRAQVATTLSQYSDLLALYQQDSEIHLQTLAQNNIAQFAQFSSEVSQLVDTIEL----RNQ- |
| 2 | 2pffB | 0.11 | 0.10 | 3.68 | 1.08 | HHsearch | | FAADDPAELVGKFLGYVNDIHALAAKLLQLVKTKELIKNYITARIMAKRPFDKKSN-SALFRAVGEGNAQLVAIFTDDYFEELRDLYQTYHVLVGDL-IKFSA-------ETLSELIR---TTLDAEKNPSNSIQLAHYVVTAKLLGELRSYLKGLVTAVIDSWESFFVSVRKAITVLFFIGV--------RCYEAYPNTSLPPSILDYVNKTNS---HLPAGKQVEISLVNGAKNLVVSGPPQSLYGLNLTLRKAKAERLPV |
| 3 | 2q13A1 | 0.06 | 0.05 | 1.98 | 0.57 | CEthreader | | ----------------------------------------------------------------MDKLPIEETLEDSPQTRSLLGVFEEDATAISNYMNQLYQAMHRIYDAQNELSAATHLTSKLLKEYVMSSTLQQFSKVIDELSSCHAVLSTQLADAMMFPITQFKERDLKEILTLKEVFQIASNDHDAAINRYSENDKVKYEVTEDVYTSRKKQHQTMMHYFCALNTLQYKKKIALLEPLLGYMQAQISFFKMGSENL-- |
| 4 | 1qoyA | 0.12 | 0.10 | 3.38 | 0.85 | EigenThreader | | ------------------------GILDSMAEIVADKTVEVVKNAIETADGALDLYNKYLDQVIQTFDETIKELVLVGDIKTLLMDSQDKYFEATQTVYEWCGVAT--------QLLAAYILL---------------FKKASAQKDILIKVLDDGIT-KLNEAQKSLLVSSQSFNNASGKLLALDSQLTNDFSAGVVEGKLIPELKNKLKSVQNFFTTLSNTVKQANKDIDAAKLKLTTEIAAIGEIKTETETLMLSLLKEA |
| 5 | 5xg2A | 0.13 | 0.11 | 3.95 | 1.23 | FFAS-3D | | ----TKGAIVRWGKRKEKLIEEIRAREEERNALVVRLGEIDRTFAVARE----------EFDTVVKELEEARK---------SLYEGEARIKRAEEEKERLKILTGEARLPGLRERAENLRRLVEEKRAEISELERRLSFELRIKLSDLEKELELA-RKDLEKVLAEERAVREEIEVAKRRINELDTLIERERGE---LAKLRGRIERL---ERKRDKLKKALENPEARELTEKIRAVEKEIAA-LREELSRVEGKLEG---- |
| 6 | 7jh5A | 0.12 | 0.12 | 4.16 | 0.88 | SPARKS-K | | ------SGSELARKLLEASTKLQRLNIRLAEALLEAIARLQELNLELVYLAVELTDPKRIRDEIKEVSKEEKEIDDAKESEKILEEAREAISELAKLLLKAIAETQDLNLRAAKAFLEAAAKLQELNIRAVELLKLTDPATIREALEHAKRRSKEIIDEAERAIRAAKRESERIIEEARRLISELARELLRAHAQLQRLELLRELLRALAQLQELNLDLLRLASEKAIARVKRESNAYYADAERLIREAAAASEKISREAERL |
| 7 | 4tqlA | 0.11 | 0.09 | 3.28 | 0.88 | CNFpred | | -------ELEKARDRMEKLYKEMVELIQKAIELMRKIFQEVKQ-----------------------------------EVEKAIEEMKKLYDEAKKKIEQMIQQIKQGDKQKMEELLKRAKEEMKKVKDKMEKLLEKLKQIMQEAKQKME----KLLKQLKEEMKKMKEKMEKLLKEMKQRMEEVK-ELLEKIKK--NIDDLKKIAEDLIKKAEENIKEAKKIAEQLVKRAKQLIEKAKQVAEELIKKILQLIEKAKEIAEKV |
| 8 | 4tqlA | 0.12 | 0.09 | 3.06 | 1.00 | DEthreader | | -------------------------------------------------------------------------EKLYKEMVELIQKAIELMRKIFQEVKQEVEKAIEEMKKLYDEAKKKIEQMIQQIKQGGDKKMEELLKRAKEEMKKVKDKMEKLLEKLKQIMQEAKQKMEKLLKQLKEEMKKMKEKMEKLLKEMKIDDLKKI-AEDLI----KKAEENIKEAKKIAEQLVKRAKQLIEKAKQVAEELIKKILQLIEKAKEE |
| 9 | 5owvC | 0.07 | 0.07 | 2.84 | 0.82 | MapAlign | | --DDSFKGRIQELCKLENELKELVFSKSSLLNLITNIEKLAFYTTTVLKEKFVKRKILNLCEILEDEVGIFDRLLNQFQYEKHLLLAYENFLKEIEILNHQILEQLKSISERISSEIFASVKEAYFYKESKGFLYTRYDYKAPYISDDAFLAMFKEFKKIKNELYKSFEEIKMKLKDFINILEREILLFKAEFSNIQKHIFQSDKNFSFCNASDYFLKDFKELLFKSILELDLFFEKLNLKFTNYENATKLSLAFFSRKINES |
| 10 | 4tqlA | 0.11 | 0.10 | 3.43 | 0.78 | MUSTER | | --------YKQMVQELEKARDRMEKLYKEMVELIQKAIELMRKIFQEVKQEV-----EKAIEEMKK---LYDEAKK--KIEQMIQQIKQG-KQKMEELLKRAKEEMKKVKDKMEKLLEKLKQIMQEAKQKMEKLLKQLKEEMKKMKEKMEKLLKEMKQRMEEVKKKMDDELLEKIKKNIDDLKKIAEDLIKKAEE--NIKEAKKIAEQLVKRAKQLIEKAKQVAEELIKKILQLIEKAKEIAEKVLKG--------------- |
| (a) | ID1 is the number of template residues identical to query divided by number of aligned residues. |
| (b) | ID2 is the number of template residues identical to query divided by query sequence length. |
| (c) | Cov is equal the number of aligned template residues divided by query sequence length. |
| (d) | Norm. Zscore is the normalized Z-score of the threading alignments. A Normalized Z-score >1 means a good alignment and is highlighted in bold. |
| (e) | Download alignment lists the threading program used to identify the template, and provide the 3D structure of aligned regions of threading templates (threading[1-10].pdb.gz). |
| (f) | Template residues identical to query sequence are highlighted in color. |
|