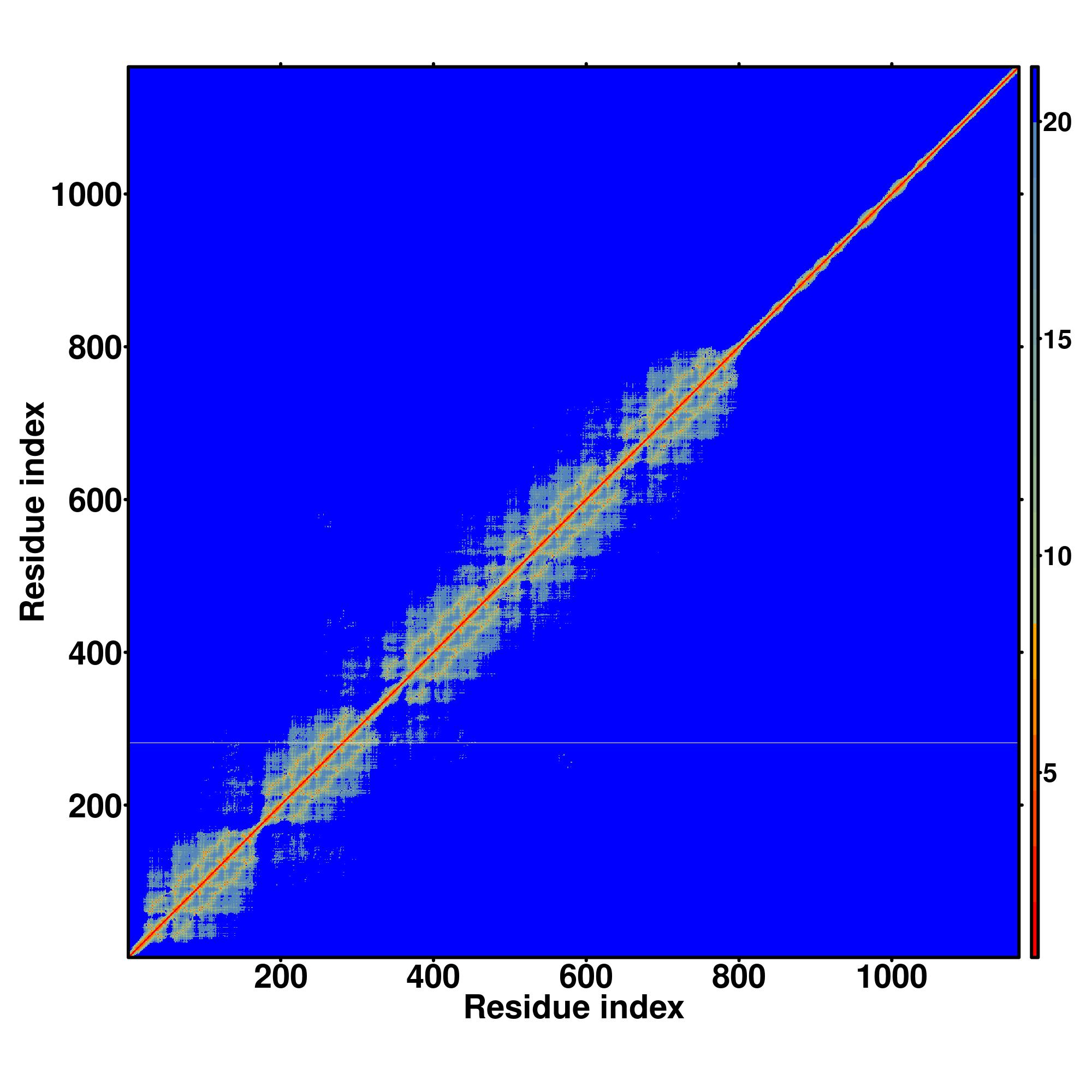
| Rank | PDB
hit | ID1 | ID2 | Cov | Norm.
Zscore | Downloadalignment | | 20 40 60 80 100 120 140 160 180 200 220 240 260 280 300 320 340
| | | | | | | | | | | | | | | | | |
|---|
| SS
Seq | CCCCCCHHHHHHHHCHHHHHHHHHHHCCCCCCCCCCCCCHHHHHHHCCCSHHHHHHHHHCCCCCCCCCCCCCHHHCCCCCCCCCCCCCCCHHHHHHCCCCCCCCCCCCCCCCCCCCCCCCCCCCHHHHHHHCHHHHHHHHHHHCCHHHHHHHHHHCCCCHHHHHHHCCHHHHHHCHHHHHHHHHHHHHHHHHHHCCCCCCCCCCCCCCCCCHHHHHHCCCCHHHHHHHHHHHHHHHHHHCCCCCCHHHHHHHHHHHHHHHHHHHHHCCCCCCCCCHHHHHHHHHHHHHHHHCCCCCCCCHHHHHCCCCCCCCSSCCCCCCCCCCHHHSSSCCCCCCCSSSSSSSSSCCCCCCCC
DKWAFTPLHEAAQKGRTQLCALLLAHGADPTLKNQEGQTPLDLVSADDVSALLTAAMPPSALPSCYKPQVLNGVRSPGATADALSSGPSSPSSLSAASSLDNLSGSFSELSSVVSSSGTEGASSLEKKEVPGVDFSITQFVRNLGLEHLMDIFEREQITLDVLVEMGHKELKEIGINAYGHRHKLIKGVERLISGQQGLNPYLTLNTSGSGTILIDLSPDDKEFQSVEEEMQSTVREHRDGGHAGGIFNRYNILKIQKVCNKKLWERYTHVHKDRSCYICHRQLLFCRVTLGKSFLQFSAMKMAHSPPGHHSVTGRPSVNGLALAEYVIYRGEQAYPEYLITYQIMRPEGMVDG |
|---|
| 1 | 5aq7A | 0.17 | 0.16 | 5.13 | 0.61 | CEthreader | | DERGHTPLHLAAYTGHLEIVEVLLKNGAGVNATDVIGTAPLHLAYKEIVEVLLKAGADVDAQDKFGKRPLWLAADQGHAEIAAKLVAARLKARVGYIELDLNSGKILESVRSEERFPMMSTFKVLLAGAILSRIDAGQEQLGRRIHYSQNDLVEYSPVTEKHLTDMTVRELASAAINTAANLLLTTIGGPKGLTAFLHNMGDHVTRLDRWEPELNEAIPNDERDTTTPVAMATTLR---------------------KLLTGELLTPASRQQLMDWMEADKVAGPLLRSVLPAGWF--IADKSGAGERGSRGIVAALGPDGKPSRIVVIYTTGSQATMDILNEAIAMLGRAMIE |
| 2 | 4oauC | 0.10 | 0.09 | 3.44 | 0.88 | EigenThreader | | KKNGATPFILAAIAGSVKLLKLFLSKGADVNECDFYGFTAFMEAAVYKALKFLYKRGANVNLRRKTKEDQERLRKGGATDVNACDNMGRNALIHALLSNVRGERGKTPLILAVEKKHRLLEQEHIEINDTDSDGKTALLLAVELKLKKIAELLCKRGASTMTARRNYD----------HSLVKVLLSHLKDLHRIYRIGKLKFFIDEKYKQEVAVKTGS-------PRAQREVSCLQSSRENSHLSESHRGHLFVCVTLTLEACLDVHRGEDVENEEDEFARNVLSSIFKAVQELHLSCGYTHQDLQPQNKSIKWAGDPQEVKRDLEDLGRLVLYVVK----------KGSISF |
| 3 | 3kr7A | 0.78 | 0.31 | 8.63 | 0.83 | FFAS-3D | | ------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------GTILIDLSPDDKEFQSVEEEMQSTVREHRDGGHAGGIFNRYNILKIQKVCNKKLWERYTHRRKEENHNHANERMLFVNAIIHKGFDESKSNQYVYGPPGHHSVTGRPSVNGLALAEYVIYRGEQAYPEYLITYQIMRPE----- |
| 4 | 5jhqA | 0.15 | 0.14 | 4.57 | 0.82 | SPARKS-K | | DDGGLIPLHNACSFGHAEVVSLLLCQGADPNARDNWNYTPLHEAAIDVCIVLLQHGADPNIRNTDGKSALDLADPSAKAVLTGEYKKDELLEAARSGNEEKLMALLTPLNVNCHASDGRKST-------------PLHLAAGY-NRVRIVQLLLQHGADVHNACSYGHELLLKHGACVNAPLHEAASVCSLLLSHGADPTLVNCHGK------SAVDMAPTPELRERLTYEFKGHSLLQAAREA-------DLAKVKKTLALEIINFKQPQSHETALHCAVTELLLRKVNEFMTPLHVAAERA------HNDVMNALDTLGQTALHRAALAGHLQTCRLLLSYGSDPSIISLQG |
| 5 | 6sa6A | 0.14 | 0.10 | 3.58 | 1.00 | CNFpred | | DETGFTPLHLAAWEGHLGIVEVLLKNGADVNANDERGHTPLHLAALEIVEVLLKNGAGVNATDVIGTAPLHLAAHLEIVEVLLKGAKAEAKDKFGKTPEDLARDNGYEAVAR---------------------LAELAALVLKLALWKLGNHASGGNLRIQWVIDAALPALVQLLSPNEQILQEALWTLGNIASGGNEQIQAVIDAG--ALPALVQLLSPNEQILQEALWTLGNIASG-------------GNEQIQAVIDAGALPALVQLL-EQILQEALWTLGNIASGQIQAVIDAGALPAL-------------------------------------------------- |
| 6 | 7jgdA | 0.08 | 0.06 | 2.23 | 0.67 | DEthreader | | DLIPLRFLQEWVENFCEQRIGIGTAGSRQIYRYHFLTTPSSYLSPWTTYTTYTTTE-KCD-TLVVVNNTYKYACQCKIPTNETCRENCKCCN--T-TGDKGKSA-DLLKENY------------------PE-ISANFDFCYIVPRQQLC-LYE-LFPIILETLQIVAEREAYYLWQAIGSFYDLEDIIKGDKLNEFGSSDTNDIDTKRRTDWWE--------------DA-QSGVRYAVEEKNENFPLCMGVIGIAKPQ-------------------NYYNKIYRKKDYSMIMA--P----------------TVIDYL-------------------GNYICC-------- |
| 7 | 5aq7A | 0.12 | 0.10 | 3.37 | 0.84 | MapAlign | | ---LGKKLLEAARAGQDDEVRILLANGADVNTADETGFTPLHLAAWEGHLGIVEVL----------------------------------------------------------------LKNGADVNANDERGHTPLHLAAYTGHLEIVEVLLKNGAGGADVDKRPLWLAAD--QGHAEIAAKLVAARLRVGYIELDLNSGKILESVRSEERFPMMSTFKVLLAGAILSRIDAAITMSDNTAANLLLTTIPNDERDTTTPVAMATTLRKLLTGELLTPASRQQLMDWMEVLPAGWFIADKSGAGERGSRGIVAALGPDGKPSRIVVIYTTGSEAIAMLGRAMIE--------- |
| 8 | 1n11A | 0.15 | 0.14 | 4.60 | 0.84 | MUSTER | | NVKVETPLHMAARAGHTEVAKYLLQNKAKVNAKAKDDQTPLHCAARIGHTNMVKLLLENNANPNLATTAGHTPLHEKEASQACMTKKGFTPLHVAAKYGKVRVAELLLERDAHPNAAGKNGPRGGSPHSPAWNGYTPLHIAAKQNQVEVARSLLQYGGSANAESVQGVTPLHLA---QEGH----AEMVALLLSKQANGN---LGNKSGLTPLHLVAQEGHVPVADVLIKHGVMVDATTRMGYT--HYGNIKLVKF--LLQHQADVNAKTKLGYSPLHQAAQQ----HTDIVTLLLKNGASPNEVSSDGTTPLAIAKRLGYISVTDVLKVTDETSFVLHRMSF----PETVDE- |
| 9 | 3kr7A | 1.00 | 0.39 | 10.92 | 1.26 | HHsearch | | ------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------GTILIDLSPDDKEFQSVEEEMQSTVREHRDGGHAGGIFNRYNILKIQKVCNKKLWERYTHVHKDRSCYICHRQLLFCRVTLGKSFLQFSAM-MAHSPPGHHSVTGRPSVNGLALAEYVIYRGEQAYPEYLITYQIMRPE----- |
| 10 | 5le2A | 0.13 | 0.13 | 4.47 | 0.54 | CEthreader | | DVIGTAPLHLAAMWGHLEIVEVLLKNGADVRAQDKFGKTPKDLANEWIRELLEKAERKLKDLDRKLLEAARAGHRDEVEDLIKNGADVNTADETGFTPLHLAAWEGHLGIVEVLLKNGADVNANDERGHTDVIGTAPLHLAAMWGHLEIVEVLLKNGADVRAQDKFGKTPKDLA---RDNGNEWIRELLEKAERKLKDLDRKLLEAARAGHRDEVEDLIKNGADVNTADETGFTPLHLAAWEGHLGIVEVLLKNGADVNANDERGHTPLHLAAYTGHLEIVEVLLKNGAGVNATDVIGTAPLHLAAMWGHLEIVNAQDKFGKTPFDLAIDNGNEDIAEVLQKAA---------- |
| (a) | ID1 is the number of template residues identical to query divided by number of aligned residues. |
| (b) | ID2 is the number of template residues identical to query divided by query sequence length. |
| (c) | Cov is equal the number of aligned template residues divided by query sequence length. |
| (d) | Norm. Zscore is the normalized Z-score of the threading alignments. A Normalized Z-score >1 means a good alignment and is highlighted in bold. |
| (e) | Download alignment lists the threading program used to identify the template, and provide the 3D structure of aligned regions of threading templates (threading[1-10].pdb.gz). |
| (f) | Template residues identical to query sequence are highlighted in color. |
|