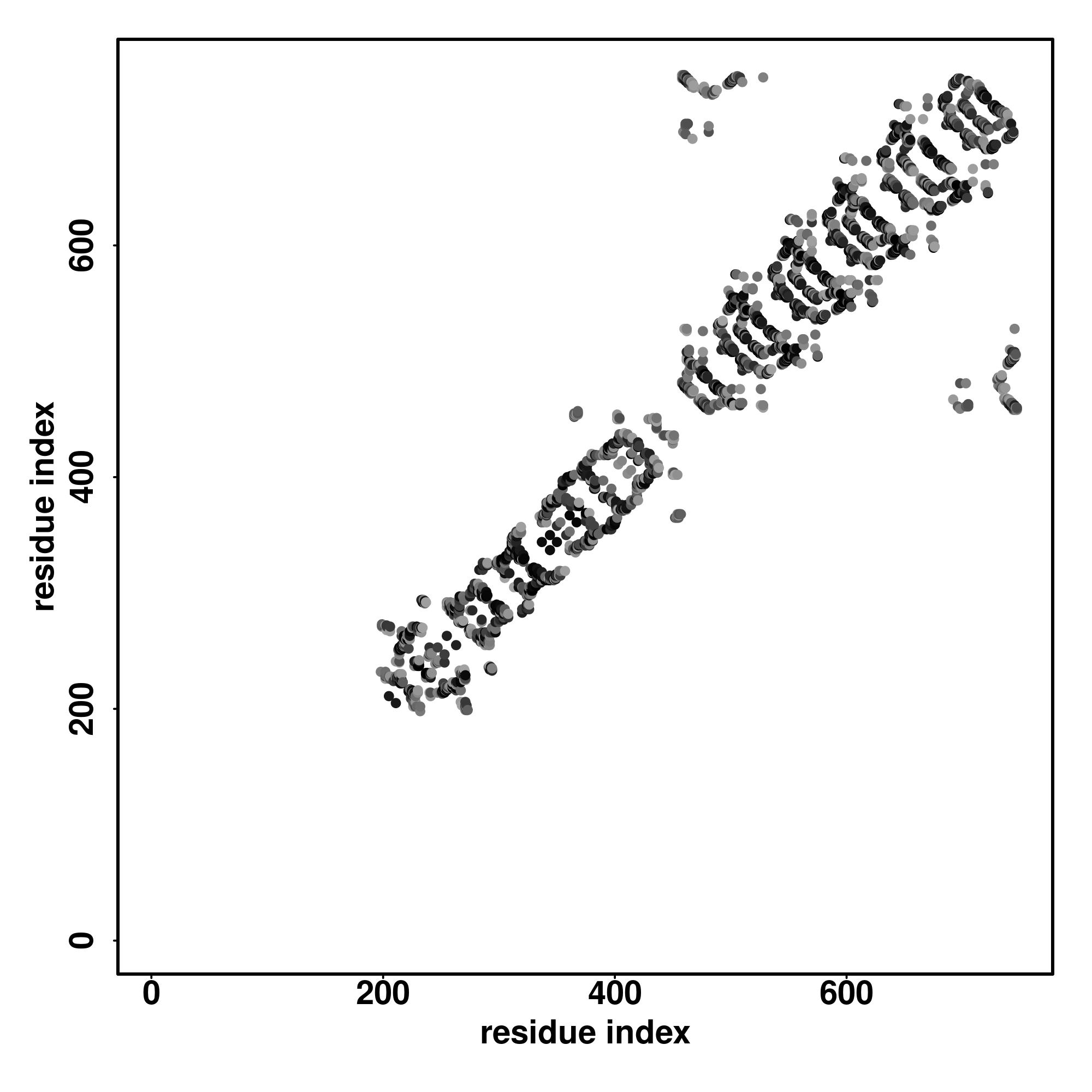
| Rank | PDB
hit | ID1 | ID2 | Cov | Norm.
Zscore | Downloadalignment | | 20 40 60 80 100 120 140 160 180
| | | | | | | | | |
|---|
| SS
Seq | CCCCCCCCCCHHHHHHHHHHHHCCCCCCCCCCCCCCSSCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCSHHHCHHHHHCCCCCCCCCCCCCCCCHHHCCCCCCCCCCCCCCCCCSSCCCCCCCCCCCCCCCCC
MSGSGRKDFDVKHILRLRWKLFSHPSPSTGGPAGGGCLQQDGSGSFEHWGPSQSRLLKSQERSGVSTFWKKPSSSSSSSSSPSSSSSSFNPLNGTLLPVATRLQQGAPGQGTQQPARTLFYVESLEEEVVPGMDFPGPHEKGLVLQELKVEPDNSSQATGEGCGHRLSSTGHSMTPQSDLDSSS |
|---|
| 1 | 1vt4I3 | 0.12 | 0.12 | 4.22 | 0.67 | CEthreader | | GGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGG-- |
| 2 | 6q4sA | 0.07 | 0.06 | 2.52 | 0.45 | EigenThreader | | FDLPFD---------------TPVSEHLPQAAARTYPHASADDMVVLMNWFSLAFLFDDQFDASRPEVARELIVTPLRPAGSPPRVACPITLAWAEVWKYLSHGMSLTWQTRFAASWG---RFLVAHCEEVDLAARGLEGTLGADTIGFMNDIHSFEREGHNLIAVLRRERGCSWQQATDEAYR |
| 3 | 7kwzA | 0.16 | 0.11 | 3.79 | 0.38 | FFAS-3D | | -----------------------------------------NPGGFGNQG------GFGNSRGGGAGLGNNQGSNMGGGMNFGAFSIN----PAMMAAAQAALQSSWGMMGMLASQQN---QSGPSGNNQNQGNMQREPNQAFGSGNNSYSGSNSGAAIGWGSASNASGSGFNGGFGSSMDSKS |
| 4 | 6ahfC1 | 0.09 | 0.08 | 2.88 | 0.99 | SPARKS-K | | -----QFTERALTILTLLASDHQHPQLQPIHIETPEDGSVPYLQNLIEKGRYDYDLFKKVVNRNLVRI------------PQQQPAPAEITPSYALGKVLQDAAKIQKQQKDSFIAQDHILFALFNDSSIQQIFKEAQVDIEAIKQQALELRGNTRIDSRGADTNTPLE--------------- |
| 5 | 3i04A2 | 0.04 | 0.03 | 1.68 | 0.67 | DEthreader | | ALEMLVSKITAFRFV-QKIGYEGIRISRGICG--ASAWT-------IVARNVGLMILTGAAAHCG------R--KFRTHN-V--VPFISILVNQAHMDNDP-------------VNLVSAIA-CGCNDSHLTVMKELLKNNVFVVVFHMGSCVDNS-AMSGKAAIGWWVLGVPTHVMPFEMDPQ |
| 6 | 1vt4I3 | 0.12 | 0.12 | 4.20 | 0.89 | MapAlign | | GGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGG----- |
| 7 | 6etxG | 0.10 | 0.10 | 3.79 | 0.76 | MUSTER | | LQGLLARWLALFLSLKASYRLHQLRSWGAPEGRNKDFLLGVNFPLSFPNLCSCPLLKSLVFSSHCKAVSGYSDQVVHQRRSATSSLRRCLLTELPSFLCVASPRVTAVPLDSYCNDRSAEYERRVLKEGGSLALLNGAPELAADWLNRRSQFFPEPAGGLWSIRPQNGWSFIRIPGKESLITDS |
| 8 | 2pffB | 0.18 | 0.16 | 5.15 | 0.59 | HHsearch | | FFVSVRKAITVLFFIGVRCEAYPNTSLPP-------SILED---SLENNELSISNLTQEQVQ----DYVNKTNSHLPA---GKQVEISLVVVSGPPQSLYGRKA---KAPSGLDQSRIPFSERKLKSNRFSHLLVPASDLINKNAKDIQIPVYDTFDGSSEDCIIRLPTTQFKATHILDFGPGG |
| 9 | 5wlcLW | 0.02 | 0.02 | 1.59 | 0.59 | CEthreader | | GQLVSELRTKAGPTMAMAQNPWNAVMHLGHSNGTVSLWSPSMPEPLVKLLSARGPVNSIAIDRSGYYMATTGADRSMKIWDIRNFKQLHSVESLPTPGTNVSISDTGLLALSRGPHVTLWKDALKLSGDSKPCFGSMGGNPHRNTPYMSHLFAENLGFVPFEDLLGVGHQTGITNLIVPGAGEA |
| 10 | 4cr2Z | 0.08 | 0.08 | 3.03 | 0.45 | EigenThreader | | SAGLDSSSFVNGFLNLG----YCNDTKGDGMTSAVASIGSIYQWNGALLGIGISASGLGIAFAGSKLLLPIAASTDLPIETAAMASLALAHVFVGTCNLGILYMGQLVGSCAYTGTGDLAYAVLGIALIALGEYGNEHIRRMVPLAMGIVSVSDPQSMNSIFAMGLCGAGTNNLAQGLLHLGKG |
| (a) | ID1 is the number of template residues identical to query divided by number of aligned residues. |
| (b) | ID2 is the number of template residues identical to query divided by query sequence length. |
| (c) | Cov is equal the number of aligned template residues divided by query sequence length. |
| (d) | Norm. Zscore is the normalized Z-score of the threading alignments. A Normalized Z-score >1 means a good alignment and is highlighted in bold. |
| (e) | Download alignment lists the threading program used to identify the template, and provide the 3D structure of aligned regions of threading templates (threading[1-10].pdb.gz). |
| (f) | Template residues identical to query sequence are highlighted in color. |
|