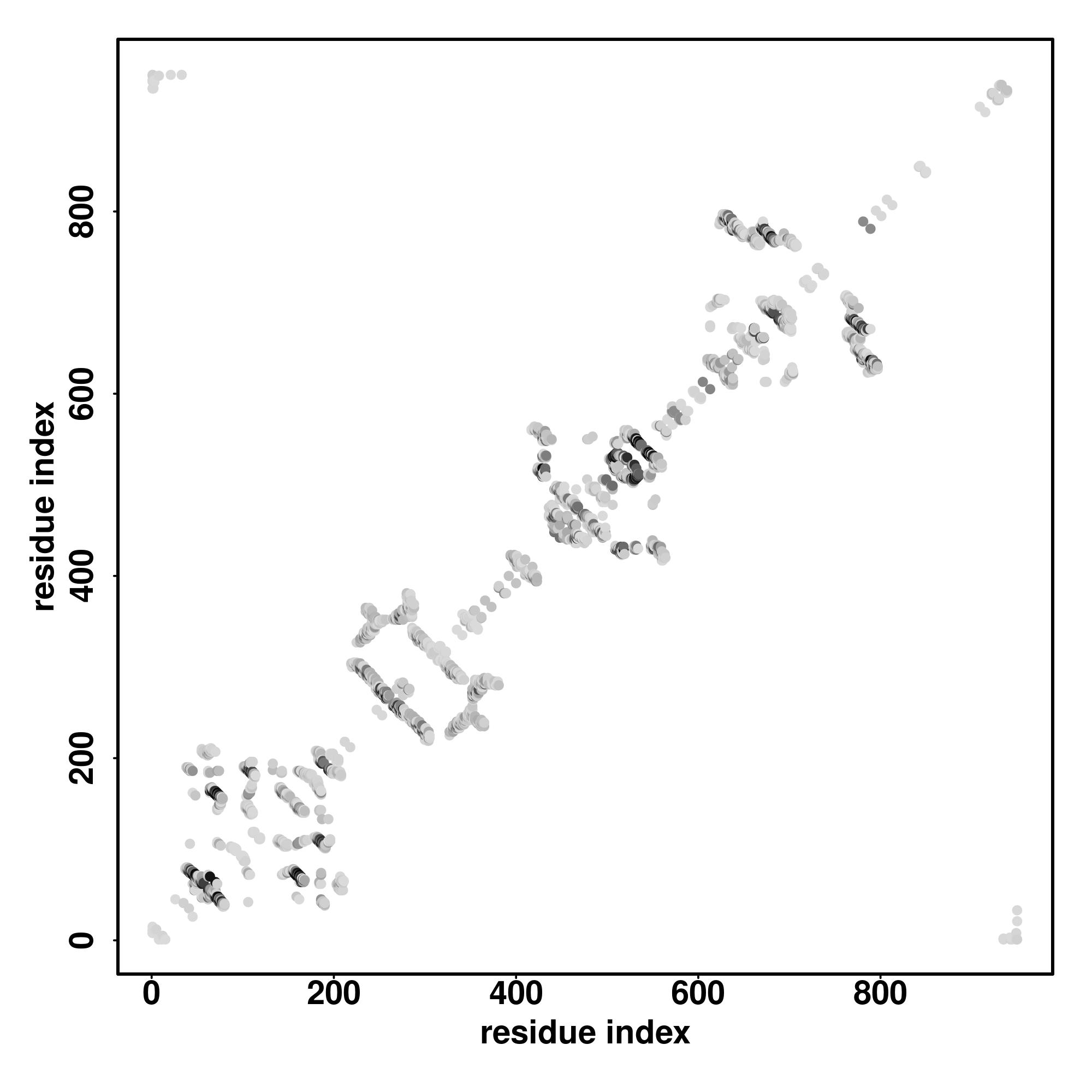
| Rank | PDB
hit | ID1 | ID2 | Cov | Norm.
Zscore | Downloadalignment | | 20 40 60 80 100 120 140 160
| | | | | | | | |
|---|
| SS
Seq | CCCHHHHHHHHCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCSHHHHHHCCHCHCCCCCCCCCCCCCSSSCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCHHHCCCCCCCCCCCCCCCHHHHHHHHHHHHHHCCCCCCCCCCCCCCCCCCCCCCCCC
CSDNDVRKVGSLAGSSVFLNRSPSRCSGLNRSESPNRERSDFGGSNTQLYSSSNITLLNNRNSLPCSRSDGLRSPSEVVYLRMEELAFTQEEMTDFEEHSTQQLTLSPAAVPTRAASDSECCNINLDTETSPCSSDFEENVGKKLLRTLSGQKRKRSPEGERTSEDNSNLTPLIT |
|---|
| 1 | 4ggaA | 0.07 | 0.07 | 2.85 | 0.56 | CEthreader | | TSSAEVQLWDVQQQKRLRNMTSHSARSGSRSGHIHHHDVRVAEHHVATLWAPDGRHLADNLVNVWPSAPGEGGWVPLQTFTQHQGAVKAVAWCPWQSNVLATGGGTSDRHIRIWNVCSGACLSAVDAHSQVCSILWSPHYKELISGHGFAQNQLVIWKYPTMAKVAELKGHTSRV |
| 2 | 1l6nA | 0.07 | 0.07 | 2.83 | 0.47 | EigenThreader | | LSGGELDKWE----KIRLKQYKLKHIVWASRELAVNPGLLETSEGCRQILGQLQPSLQTGSYNTIAVLYCVHQRIDVKDTKEALDKIEEEQNKSKKKAQQAAADTGNVGGHQAAMQMLKETINEEAAEWDRLHPVEPRGSDIAGTTSTLQEQIGWMTHNPPIPVGEIYKRWIILG |
| 3 | 2xd8A | 0.06 | 0.06 | 2.45 | 0.35 | FFAS-3D | | -----------MANA------NQVALGRSNLSTGTGYGGATDKYALYLKLFSGEMFKGFQHETIARDLVTKRTKNGKSLQFIYTGRMTSSFHTPGTPILGNADKAPPVAEKTIVMDDETLAHYELRGEISKKIGYALAEKYDRLIFRSITRGARSASPVSATNFVEPGGTQIRVG |
| 4 | 5yfpH2 | 0.11 | 0.11 | 3.89 | 0.96 | SPARKS-K | | RDRSSVLILEKFWDTELDQLFKNVEGAQKFINSTKGRHILMNSANWMELNTTTQIFILND-LVLIADKSRDKQNDF------IVSQCYPLKDVTVTQEEFSTKKFSNSNSSLYECRDADECIRKAKDDLCDIFHVEEENKRIRESFRYLQSTQQTPGRENNRSPNKNKRRSMGGS |
| 5 | 5mqhA | 0.02 | 0.01 | 0.44 | 0.35 | CNFpred | | ----------------------------------------------------------------------------DLLIMMSDGIFEGPKHVENHDLW--------------------------MKRKMKGLKTNDPQEIADLLMEEVIRT----------------------- |
| 6 | 5lj3T | 0.02 | 0.02 | 1.21 | 0.67 | DEthreader | | ---------------------ETIFILSYLAIRHALASSLMMEMHRKVWDPVIKFVEEKV---E---------STDEAELVLILRNIKGAKIGRDSCGDLLKLQEKCNVSYKLWINWADREKYYLEYENSEEVATPIRLFEKALKNLCS-----------SKIEILRRGAGESRQ |
| 7 | 2j04A | 0.09 | 0.09 | 3.28 | 0.82 | MapAlign | | LSNNGNVSVFKDNKMLTNLSSIVVGNEDGELQFFSIRKNSENTPEFYVWYEDVLVAALSNNSVFSMTVSASSHQPVSRMIQNASRRKITDLKIVDYKVVEMRIIMFLNMTIDKPSILSFLEALYEYAINKKSELTNSFDLACVLSIAAILKREAKRCGVTLLPILTTHV------ |
| 8 | 4k0mC | 0.10 | 0.09 | 3.38 | 0.60 | MUSTER | | RSDQNVRGTVSL---PHGLGKQ-VRVLAIAKGEKI-KEAEEAGADYVGGEEIIQAAAAAAAAAAAAAAAAAAAAAA----------AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA |
| 9 | 2pffB | 0.19 | 0.18 | 5.90 | 0.77 | HHsearch | | SFFVSVRKNTSLPPSILLENNEGVPSPMLSISNLTQEQVQDYVNKTNHLPAGKQISLVNGAKNLVVSGPPTLRKAKAPSGLDQSRIPFSERKL-KFSNRFLPVASHSHLLVPAS------DLINKDLVKNNVSQIPVYDTFDGSDLRVLSGSIRLPVKWETFTLDINPDDDYGFK |
| 10 | 1p22A | 0.11 | 0.11 | 3.93 | 0.48 | CEthreader | | QYDERVIITGSSDSTVRVWDVNTGEMLNTLIHHCEAVLHLRFNNGMMVTCSKDRSIAVWLRRVLVGHRAAVNVSASGDRTIKVWNTS-TCEFVRTLNGHKRGIACLQYRDRLVVSGSSDNTIRLWDIECGACLRVLEGHEELVRCIRFDNKRIVSGAYDGKIKVWDLVAALDPRA |
| (a) | ID1 is the number of template residues identical to query divided by number of aligned residues. |
| (b) | ID2 is the number of template residues identical to query divided by query sequence length. |
| (c) | Cov is equal the number of aligned template residues divided by query sequence length. |
| (d) | Norm. Zscore is the normalized Z-score of the threading alignments. A Normalized Z-score >1 means a good alignment and is highlighted in bold. |
| (e) | Download alignment lists the threading program used to identify the template, and provide the 3D structure of aligned regions of threading templates (threading[1-10].pdb.gz). |
| (f) | Template residues identical to query sequence are highlighted in color. |
|