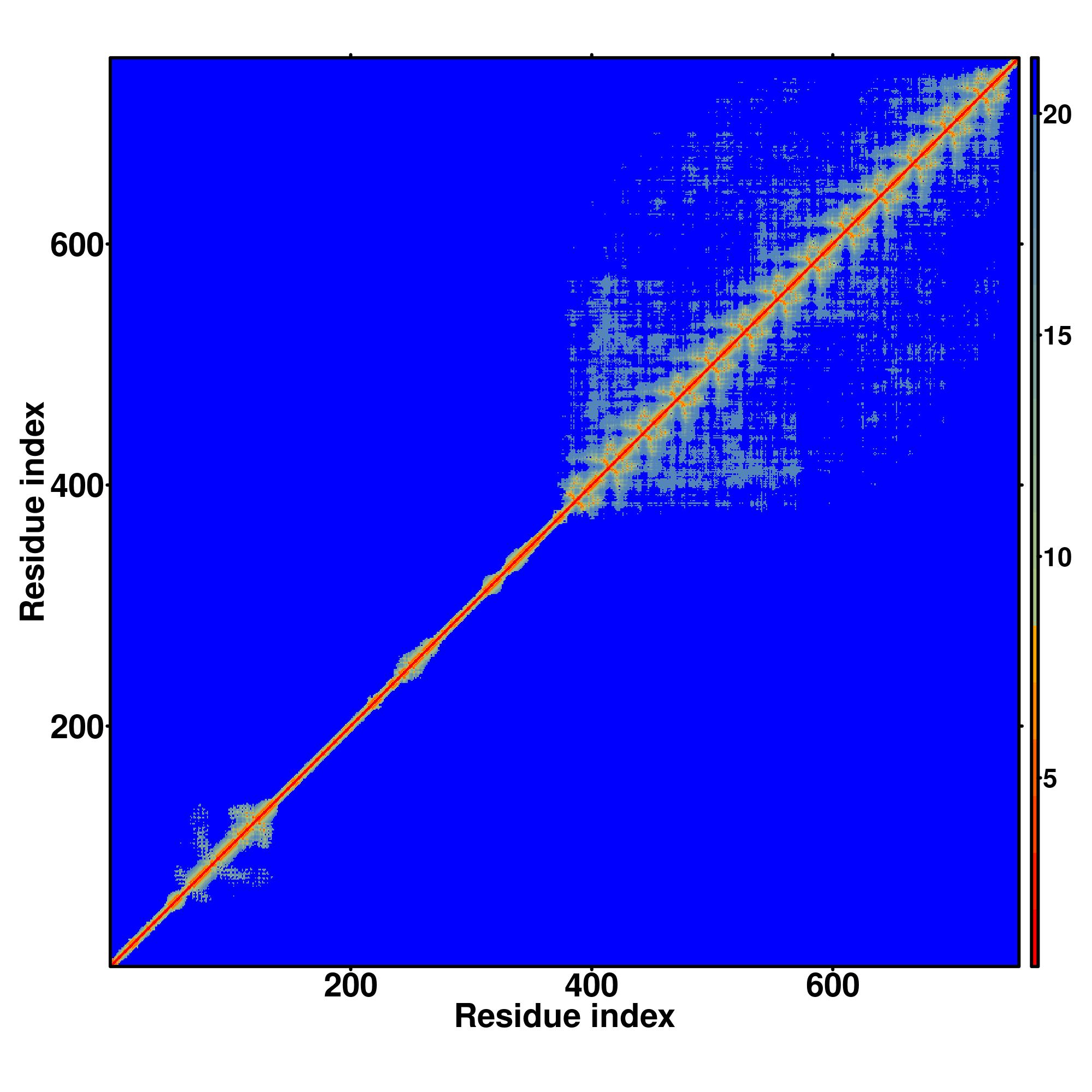
| Rank | PDB
hit | ID1 | ID2 | Cov | Norm.
Zscore | Downloadalignment | | 20 40 60 80 100 120 140 160 180 200 220 240 260
| | | | | | | | | | | | | |
|---|
| SS
Seq | CCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCSSSSSSSSSCCHHHHHCCCHHHHHHHHHHHHHHHHCHHSSCCCCCCCCSCCCSCCCCCCCCCCCCSSCCCCCCCCCCCCCCCCCHHHHCSSSCCCCCCCCCCCCCCCCHHHCCCCCCCCCCCCCCCCCCCCCSSCCCCCCHHHCCCSCCCCCSSCCCCCCSSCCCCCCCCSSCCSCCCCCSCCCCCCCSSCCCCCHHHSSCCCCCCCC
SPLSGGSAPGAHLEPPYDPGTHHLPSGDFAQCTSPVPTLPQVGNSGDQAGATVLRMVRPQDTVAYEDLSVDYTQKKWKSLTLSQRALQWNMMPENHHSMASLAGENMMKGSELTPKQEFFKGSESSNRTSGGLFGVVPGAAETGDVCEDTFKELEGQTSDEEGSRLENDFLEITDEDKKKSTKDRYDKYKEVGEHPPLSSSPVEHEGVLKGQKSYRCDECGKAFNRSSHLIGHQRIHTGEKPYECNECGKTFRQTSQLIVHLRTHTGEKP |
|---|
| 1 | 5v3jE | 0.18 | 0.17 | 5.54 | 0.80 | CEthreader | | ------------PHKCKECGKAFHTPSQLSHHQKLHVGEKPYKCQECGKAFPSNAQLSLHHRVHTDEKCFECKECGKAFMRPSHLLRHQRIHTGEKPHKCKECGKAFRYDTQLSLHLLTHAGARRFECKDCDKVYSCASQLALHQMSHTGEKPHKCKECGKGFISDSHLLRHQSARHQRAHSGDKPYKCKECGKSFTCTTELFRHQKVHTGDRPHKCKECGKAFIRRSELTHHERSHSGEKPYECKECGKTFGRGSELSRHQKIHT---- |
| 2 | 7kdtA | 0.03 | 0.03 | 1.86 | 0.58 | EigenThreader | | NKYFKAGKYEQAIQCYTEAISLCPTEKNVDLSTFYQNRAAAFEQLQKWKEVAQDCTKAVELNP---------KYECLEDVTAVCILEIISECSKEIDAE---GKYMAEALLLRATFYLLIGNANAAKPDLDKVISLKEANVKLRANALIKRGSMYMQQQQPLLSTQDFNMAADIDPQNADVYHHRGQLKILLDQVEEAVADFDECIRLRPESALAQAQKCFALYRNNQAAMKGFEEVIKKFPRCAEGYALYAQALTDQYDKCIDLEPDNA |
| 3 | 5v3jE1 | 0.40 | 0.16 | 4.81 | 1.35 | FFAS-3D | | ---------------------------------------------------------------------------------------------------------------------------------------------------------------PHKCKECGKAFPSQLSHHQKLHVGEKPYKCQECGKAFPSNAQLSLHHRVHTDEKCFECKECGKAFMRPSHLLRHQRIHTGEKPHKCKECGKAFRYDTQLSLHLLTHAGAR- |
| 4 | 1hn0A | 0.05 | 0.04 | 1.80 | 0.83 | DEthreader | | --------KNLMQSLIVPTKEASAWGRVDDAYQKQIIIYQ------LNSQDKQLFDNYVILGNYT-TLMFNISRAYVLEKDPTQKAQLKQMYLLMTKHLLDQ-GFVKGS--YISTLLMSDA--------QTQVYDLLSFTLSRQHLALLLL-PD--DQK---RINL-VNTFSHYITALASGNLKKAMVSAWIYSNGLPQG---F--YAF-NGGAFYNTERGFSGTSGMMA-FNLIYPPNFTNIN------KDASYEY-QVRK-------- |
| 5 | 2i13A | 0.28 | 0.15 | 4.64 | 2.50 | SPARKS-K | | ------------------------------------------------------------------------------------------------------------FSRSDHLAEHQRTHKPYKCPECGKSFSDK-------KDLTRHQRTHTGEKPY-KCPECGKSFS--QRANQRTHTGEKPYACPECGKSFSQLAHLRAHQRTHTGEKPYKCPECGKSFSREDNLHTHQRTHTGEKPYKCPECGKSFSRRDALNVHQRTH----- |
| 6 | 5v3jE | 0.16 | 0.15 | 5.04 | 1.13 | MapAlign | | ------------PHKCKECGKAFHTPSQLSHHQKLHVGEKPYKCQECGKAFPSNAQLSLHHRVHTDEKCFECKECGKAFMRPSHLLRH-QRIHTGEKPHKCKECGKAFRYDTQLSLHLLTHAGARRFECKDCDKVY----SCASQLALHQMSHTGEKPHKCKECGKGFISDSHLLRHQSVHTGETPYKCKECGKGFRRGSELARHQRAHSGDKPYKCKECGKSFTCTTELFRHQKVHTGDRPHKCKECGKAFIRRSELTHHERSHSGEKP |
| 7 | 5v3gA | 0.40 | 0.17 | 4.91 | 1.97 | CNFpred | | ---------------------------------------------------------------------------------------------------------------------------------------------YVCRECGRGFRDKSHLLS-----------------HQRTHTGEKPYVCRECGRGFRDKSNLLSHQRTHTGEKPYVCRECGRGFSWQSVLLRHQRTHTGEKPYVCRECGRGFRDKSNLLSHQRTHTGEKP |
| 8 | 5v3jE | 0.22 | 0.18 | 5.72 | 1.35 | MUSTER | | ---------------------------------PHKCKECGKAFHTPSQLSHHQKL----------------VGEKPYKCQECGKAFPSNAQLSLHHRVHTDEKCFECKEQRIHTGEKPHKCKECGKAFRYDTQLSLHLLTHAGAECKALHQMSHTGEKPHKCKECGKGFISDSHRHQSVHTGETPYKCKECGKGFRRGSELARHQRAHSGDKPYKCKECGKSFTCTTELFRHQKVHTGDRPHKCKECGKAFIRRSELTHHERSHSGEKP |
| 9 | 1x6fA | 0.18 | 0.05 | 1.68 | 1.22 | HHsearch | | ----------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------GSSGSSGLKRDFIILGGPRLQNSTYQCKHCDSKLQSTAELTSHLNIHNEE----FQKRAKRQERRKQLLSKQKYADGAFA |
| 10 | 1vt4I3 | 0.09 | 0.09 | 3.40 | 1.08 | MapAlign | | HYNIPKTFDSDDLIPPYLDQYFYSHIGHHLKNIEHPERMTLFRMVFLDFRFLEQKIWNASGSILNTLQQLKFYKPYICDNDPKYERLVNAILDFLPGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGG |
| (a) | ID1 is the number of template residues identical to query divided by number of aligned residues. |
| (b) | ID2 is the number of template residues identical to query divided by query sequence length. |
| (c) | Cov is equal the number of aligned template residues divided by query sequence length. |
| (d) | Norm. Zscore is the normalized Z-score of the threading alignments. A Normalized Z-score >1 means a good alignment and is highlighted in bold. |
| (e) | Download alignment lists the threading program used to identify the template, and provide the 3D structure of aligned regions of threading templates (threading[1-10].pdb.gz). |
| (f) | Template residues identical to query sequence are highlighted in color. |
|