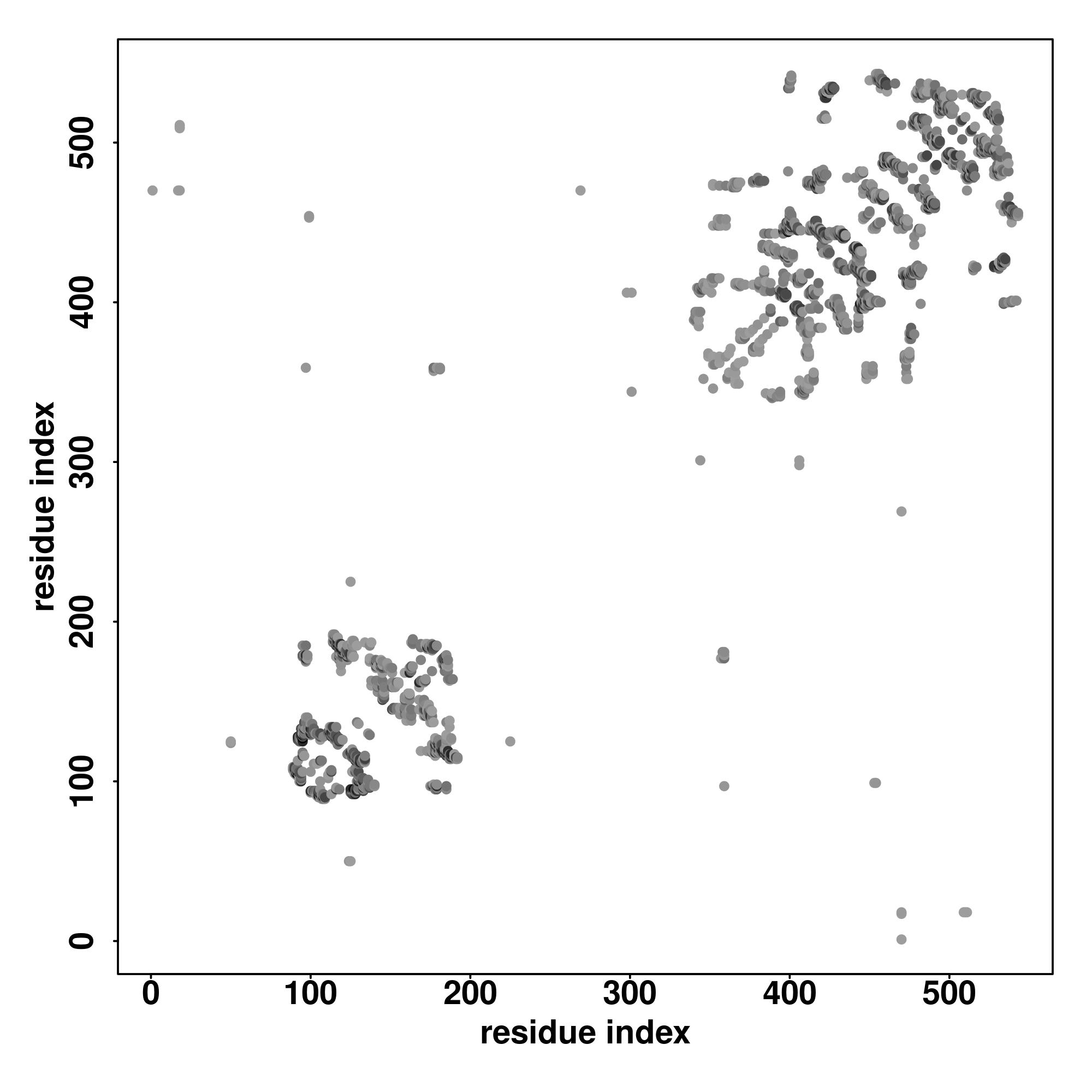
| Rank | PDB
hit | ID1 | ID2 | Cov | Norm.
Zscore | Downloadalignment | | 20 40 60 80 100 120 140 160 180
| | | | | | | | | |
|---|
| SS
Seq | CCCCCCCCCCCCCCCCCCCCCHHHCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCHHHCSSSSSCCCCCCCHHHHHHHHHHHHHHHHHHHHHHHCCCCCCCHHHHHHHHHHHHHHHHHHHHHCCCCCCC
MPNPKNSKGGRKNKRANSSGDEQENGAGALAAAGAAGAAAGGALAAAAGCGAAAAGAPGAGGAAGAGGAGTGAANAAAAAGAAAAGDAKNEAPCATPLICSFGRPVDLEKDDYQKVVCNNEHCPCSTWMHLQCFYEWESSILVQFNCIGRARSWNEKQCRQNMWTKKGYDLAFRFCSCRCGQG |
|---|
| 1 | 1ru3A | 0.08 | 0.08 | 3.21 | 0.44 | CEthreader | | LPVIRAFSGEEVRTLKDMVPILNRMRAQIKSELTFENARLAGEATWYAAEIIEALRYLKHTPENPIVVPPWTGFIGDPVVRQYGIKMVDWTIPGEAIIIGRAKDSKAAKKGKGLMLFLCDEIIIAYPLGNFTQVVHAANYALRAGLMFGGIAPGLRDAHRDYQRRRVLGEHDMVKTAAAMGAI |
| 2 | 5m45A | 0.06 | 0.05 | 2.22 | 0.68 | EigenThreader | | VRTTRYWMLDERTRIAGCHMIRKLVEEVVAEEGIEAYWKFAYEAVEHGRLGLQARIKAMTLIPSEMINDGAAYGLVSAWTALWRGLSEVNAGNANNGTGAGLSHAAAIWNPEGDQIKGGSGKYRGGCGDW-------------------GLMGGYPELIASGFSDYDLYLNYM-RGGPGFGDP |
| 3 | 6sehC2 | 0.16 | 0.07 | 2.36 | 0.44 | FFAS-3D | | ------------------------------------------------------------------------EPIKDYVAKGQEIFEFERQGACVV---CRE----EMASGDGLQALCTNQGC--DGVGHLSCWSRHWGNMMKELTL----RTRGQKEVEKLL--KR---------------- |
| 4 | 3jc8Oa | 0.08 | 0.07 | 2.80 | 0.90 | SPARKS-K | | VGALTAANFFMVIQPTEEEIGWAVAERRKLDLELADKSEIAQNLNERRREMDVLEQKLSEALTELPEQRDIEELLAQINDIGKKSGELSSVTPG---------KESVGGGEFFIKMTVSGN-------------YHEIALFLQEMANMRRIVNVNNIKFDSAKLKNEKVVLQSEFQATTFRFV |
| 5 | 2v9vA | 0.11 | 0.05 | 1.79 | 0.55 | CNFpred | | ---------------------------EAATRASLSLEETRKLLQSMAA------------------------------------------------------------AGQVTLLRVEN----DLYAISTERYQAWWQAVTRALEEFHSR-GLAREELRSRY-PARVYQALLEEWS------ |
| 6 | 1kpkA | 0.07 | 0.06 | 2.48 | 0.83 | DEthreader | | ------LRRRQLQLRDKTPLIL-MAAVVGTLVGLAAVAFDKGVAWL-QNQRMGALVHT----PLLLTVAFLCSAVLAMFGYFLVRYAPEAGSGIP-LGMVL------I-GIFGFVTLLCFSGAPGG-IFA--PMLALGTVLGTAFGMVAVLFYLEAGTFAIAGIMIITGLAT-AGPLYI---T |
| 7 | 4hvcA1 | 0.08 | 0.08 | 3.21 | 0.68 | MapAlign | | AKKEENLADWYSQVITKSEMIEYHLRPWAYAIWEAIKDFFDAEIKKGVEPEVAWVTRSGKTELAEPIAIRPTSETVMYPAYAKWVQSHRDLPIKLNQWCNVVRWEHPQPFLRTREFLWQEGHSAFATMEAAEEVLQILDLYAQVYELLAIPVVKGRKDYTTTIEAFIQGGTSHKQFAYQNSWG |
| 8 | 4k0mC | 0.33 | 0.23 | 6.89 | 0.59 | MUSTER | | EEAGADYVGGEEIIQAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA-------------------------------------------------------- |
| 9 | 2v86A | 0.16 | 0.07 | 2.19 | 0.61 | HHsearch | | ---------------------------------------------------------------------------------GPLGSPEFGYWITCCPT-CDVDVPFYSELNKPAMIYCSHG---DGHWVHAQCMDLEER-TLIHLSEGSNK-YYCNEH----------VQIARA--------- |
| 10 | 5tprA2 | 0.08 | 0.07 | 2.86 | 0.43 | CEthreader | | ----------DFSLLRTLPTDQVRNGMAELVKIAVVAHQEVFELLEKYGEELLRTHFGNIDATPEIKEIAHRLTYKAIHKMLELEVPNLHELDLDRVIAYGHTWSPTLELAPRVDMAFSATIAARRGYITIAERDRILGLMSRVGLSLDHPMLDIDILWRGTESITLTRDGLLRAAMPKPIGD |
| (a) | ID1 is the number of template residues identical to query divided by number of aligned residues. |
| (b) | ID2 is the number of template residues identical to query divided by query sequence length. |
| (c) | Cov is equal the number of aligned template residues divided by query sequence length. |
| (d) | Norm. Zscore is the normalized Z-score of the threading alignments. A Normalized Z-score >1 means a good alignment and is highlighted in bold. |
| (e) | Download alignment lists the threading program used to identify the template, and provide the 3D structure of aligned regions of threading templates (threading[1-10].pdb.gz). |
| (f) | Template residues identical to query sequence are highlighted in color. |
|