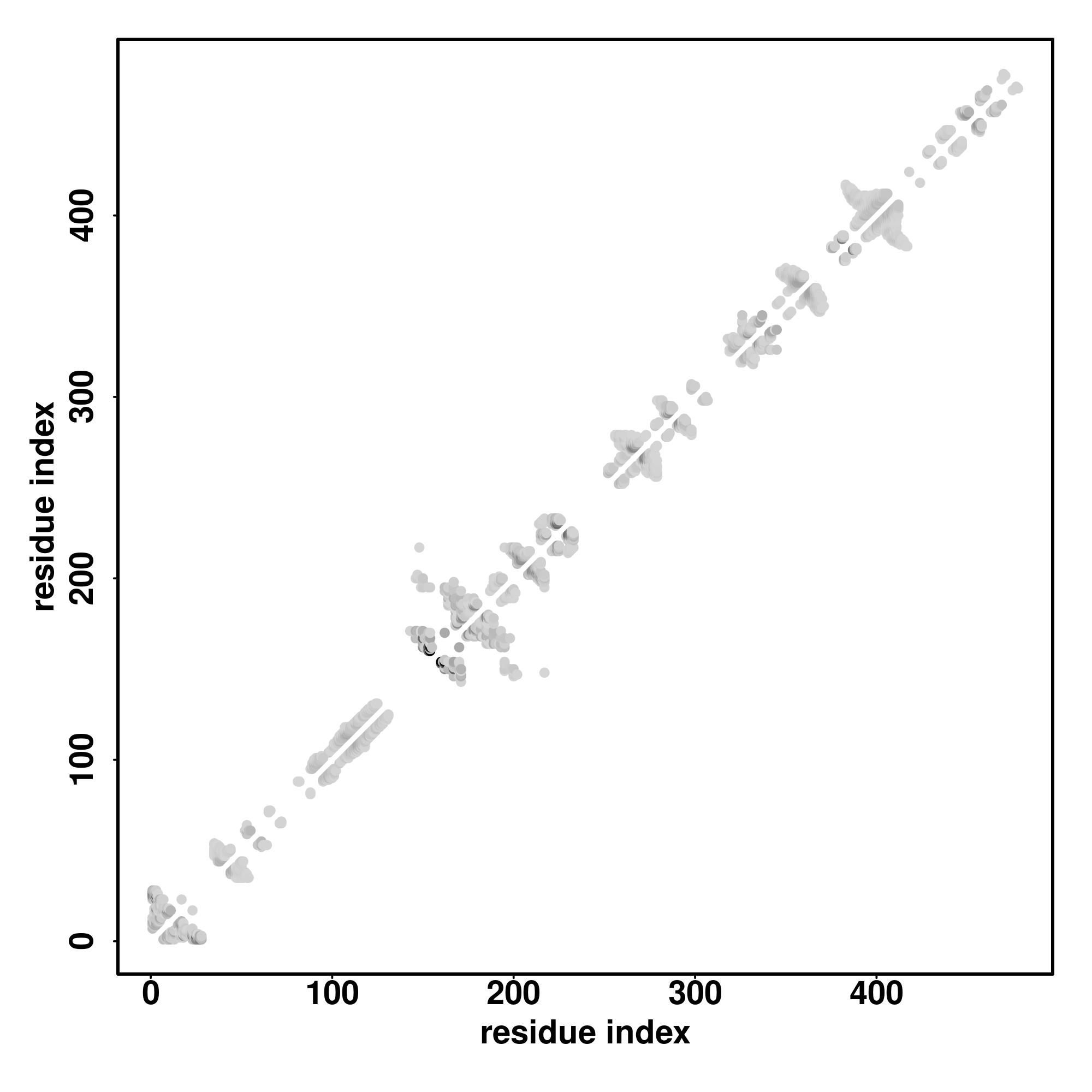
| Rank | PDB
hit | ID1 | ID2 | Cov | Norm.
Zscore | Downloadalignment | | 20 40 60 80 100 120 140 160
| | | | | | | | |
|---|
| SS
Seq | CCHHHHHHHHHCHHHHHHHHHCCCCCCCCCCCCCCCCCCCCCCCCCCCHHHHHHHHCCCCCCCCCCCCCCCCCCCCCHHHHCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCHHHHHCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCC
REQTALMMQSFGAYSLAAAAGAAGPYGRPYGLHPAAAAGAYSHPAAAAAAAAAAALQYPYALPPVAPVLPPAVPLLPSGELGRKAAAFGSQLGPGLQLQLNSLGAAAAAAGTAGAAGTTASLIKSEPSARPSFSIENIIGGGPAAPGGSAVGAGVAGGTGGSGGGSTAQSFLRPPGTV |
|---|
| 1 | 5a1uC | 0.07 | 0.07 | 2.82 | 0.48 | CEthreader | | NESKAWEVDTCRGHYNNVSCAVFHPRQELILSNSEDKSIRVWDMSKRTGVQTFRRDHDRFWVLAAHPNLNLFAAGHDGGMIVFKLERERPAYAVHGNMLHYVKDRFLRQLDFNSSKDVAVMQLRSGSKFPVFNMSNAVLLCTRASNLENSTYDLYTIPKDADSQNPDAPEGKRSSGLT |
| 2 | 5t77A | 0.04 | 0.04 | 2.04 | 0.50 | EigenThreader | | SPSIYFIFLWAISYSILNTN---NKFFWPALTPSISNITIIIGTFLSPTIGFLIGSILMFFSIIKSIIKGAFGLPFYGIYSTISRSYHAIKNTKTPFIAATIVSLSNIILDIIFGLKYGVALATSIAGIIGVLYLLFSVKTFPFVIYLTDFTDNEFWFLIQILIGILVYLIFSSIFYR |
| 3 | 6j94A1 | 0.13 | 0.09 | 3.11 | 0.39 | FFAS-3D | | -E----MESLFSRLTLDIIGKAV------FNYDFDSLTNDTGVIEAVYTVLREAEDRS---------VSPIPVWDIPIWKDISPRQRKVATSLKLINDTLDDLIATCKRMVEEEELQFHEEYMNERDPSGDDVSSKQLRGH------------------------------------- |
| 4 | 6ahfC1 | 0.11 | 0.09 | 3.26 | 1.09 | SPARKS-K | | TERALTILTLAQKLA----SDHQHPQLQPIHILAAFIETPEDG--------SVPYLQNLIEKG---RYDYDLFKKVVNRNLVRIPQQQPAPAEITPSYALGKVLQDAAKIQKQQKDSFFNDSSIQQIFKEAQVDIEAIKQQALELRGNTRIDSRGADTNTPLE--------------- |
| 5 | 4kqjA | 0.10 | 0.03 | 1.03 | 0.59 | CNFpred | | PRDGIDVLSKVGGFDLVGMTGVMLGAACGLPVLLDGFL-----------SYSAALAACQ----------------------------------------------------------------------------------------------------------------------- |
| 6 | 2vcaA | 0.09 | 0.07 | 2.60 | 0.83 | DEthreader | | NK--YNPIMLTYSYT----------------------VLDFAYMQGYGVQLDLFNMLHNDAAPELRWIIFEAIEIFAYDFKDS-DAFLYDFADILKQLLANSAQEYMCNANGNGEK-FKFVSGKFLELIKQERVLSTRPE---GNWINARALTTWGSRNAKDYSNRQW---------- |
| 7 | 6ybt1 | 0.04 | 0.04 | 2.21 | 0.84 | MapAlign | | PAHAVDAVKNATFHQRGIALWGGEKFKQIQRFSHQGVQLIDFSPCERYLVTFSKKRGFHCESSAHWPIFKWSHDGKFFARMTLDTLSIYETPSMGLLDKKSLKISGIKDFSWSPGGNIIAFWVPEDKDIPARVTLMQLPTRQEQKNGDYLCVKVDRVVTNFEIFRMREKQVPVDVVEM |
| 8 | 4k0mC | 0.18 | 0.18 | 5.85 | 0.96 | MUSTER | | FDETVEVHAKLGIDPRRSDQNVRGTVSLPHGLGKQVRVLAIAKGEKIKEAEEAGADYVGGEEIIQAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA- |
| 9 | 5it7X | 0.15 | 0.10 | 3.39 | 0.45 | HHsearch | | GLNSARKLRELGTAFKSSPF-GGSSH--AKGIVLEKIGIESK-------QPNSAI-------RKCV-----RVQLI-KN--GKKVTAFVPNDGC-----LNFVDE---------------------------NDEVLLAGFGRKGKAKDIPG--VRF-KVVKVSGVSLLALWKEKKEK |
| 10 | 3fvhA | 0.09 | 0.09 | 3.41 | 0.44 | CEthreader | | VRQEEAEDPACIPIFWVSKWVGYQLCDNSVGVLFNDSTRLILYNDGDSLQYIERDGTESYLTVSSHPNSLMKKITLL-KYFRNYMSEHLLKAGANITPREGDELARLPYLRTWFRTRSAIILHLSNGSVQINFFQDHT--KLILCPLMAAVTYIDEKRDFRTYRLSLLEEYGCCKELA |
| (a) | ID1 is the number of template residues identical to query divided by number of aligned residues. |
| (b) | ID2 is the number of template residues identical to query divided by query sequence length. |
| (c) | Cov is equal the number of aligned template residues divided by query sequence length. |
| (d) | Norm. Zscore is the normalized Z-score of the threading alignments. A Normalized Z-score >1 means a good alignment and is highlighted in bold. |
| (e) | Download alignment lists the threading program used to identify the template, and provide the 3D structure of aligned regions of threading templates (threading[1-10].pdb.gz). |
| (f) | Template residues identical to query sequence are highlighted in color. |
|